false2026Q10001655759--12-319111xbrli:sharesiso4217:USDiso4217:USDxbrli:sharesxbrli:purearvn:productutr:Harvn:segment00016557592026-01-012026-03-3100016557592026-05-0700016557592026-03-3100016557592025-12-3100016557592025-01-012025-03-310001655759us-gaap:CommonStockMember2025-12-310001655759us-gaap:RetainedEarningsMember2025-12-310001655759us-gaap:AdditionalPaidInCapitalMember2025-12-310001655759us-gaap:AccumulatedOtherComprehensiveIncomeMember2025-12-310001655759us-gaap:TreasuryStockCommonMember2025-12-310001655759us-gaap:AdditionalPaidInCapitalMember2026-01-012026-03-310001655759us-gaap:RetainedEarningsMember2026-01-012026-03-310001655759us-gaap:CommonStockMember2026-01-012026-03-310001655759us-gaap:AccumulatedOtherComprehensiveIncomeMember2026-01-012026-03-310001655759us-gaap:CommonStockMember2026-03-310001655759us-gaap:RetainedEarningsMember2026-03-310001655759us-gaap:AdditionalPaidInCapitalMember2026-03-310001655759us-gaap:AccumulatedOtherComprehensiveIncomeMember2026-03-310001655759us-gaap:TreasuryStockCommonMember2026-03-310001655759us-gaap:CommonStockMember2024-12-310001655759us-gaap:RetainedEarningsMember2024-12-310001655759us-gaap:AdditionalPaidInCapitalMember2024-12-310001655759us-gaap:AccumulatedOtherComprehensiveIncomeMember2024-12-310001655759us-gaap:TreasuryStockCommonMember2024-12-3100016557592024-12-310001655759us-gaap:AdditionalPaidInCapitalMember2025-01-012025-03-310001655759us-gaap:RetainedEarningsMember2025-01-012025-03-310001655759us-gaap:CommonStockMember2025-01-012025-03-310001655759us-gaap:AccumulatedOtherComprehensiveIncomeMember2025-01-012025-03-310001655759us-gaap:CommonStockMember2025-03-310001655759us-gaap:RetainedEarningsMember2025-03-310001655759us-gaap:AdditionalPaidInCapitalMember2025-03-310001655759us-gaap:AccumulatedOtherComprehensiveIncomeMember2025-03-310001655759us-gaap:TreasuryStockCommonMember2025-03-3100016557592025-03-310001655759arvn:PfizerInc.Memberarvn:CollaborationAgreementMember2021-07-310001655759arvn:RegulatoryAndSalesBasedMilestonesMemberarvn:PfizerInc.Membersrt:MaximumMember2021-07-310001655759arvn:PfizerInc.Memberarvn:RegulatoryMilestonePaymentsMember2021-07-310001655759arvn:PfizerInc.Memberarvn:SalesBasedMilestonesMember2021-07-310001655759arvn:PfizerInc.Memberarvn:CollaborationAgreementMember2026-01-012026-03-310001655759arvn:PfizerInc.Memberarvn:CollaborationAgreementMember2026-03-310001655759arvn:PfizerInc.Memberarvn:DevelopmentMilestonePaymentsMember2024-06-180001655759arvn:PfizerInc.Member2018-01-012018-12-310001655759arvn:OptionPaymentsToLicenseAgreementMemberarvn:PfizerInc.Membersrt:MaximumMember2026-03-310001655759arvn:DevelopmentMilestonePaymentsMemberarvn:PfizerInc.Membersrt:MaximumMember2017-12-310001655759arvn:SalesBasedMilestonePaymentsMemberarvn:PfizerInc.Membersrt:MaximumMember2017-12-310001655759arvn:NovartisPharmaAGMemberus-gaap:LicenseMember2024-05-310001655759arvn:GenentechIncorporationAndFHoffmanLaRocheLimitedMember2017-11-012017-11-300001655759arvn:GenentechIncorporationAndFHoffmanLaRocheLimitedMember2015-09-012015-12-310001655759arvn:DevelopmentMilestonePaymentsMemberarvn:GenentechIncorporationAndFHoffmanLaRocheLimitedMembersrt:MaximumMember2026-03-310001655759arvn:GenentechIncorporationAndFHoffmanLaRocheLimitedMemberarvn:RegulatoryMilestonePaymentsMember2026-03-310001655759arvn:GenentechIncorporationAndFHoffmanLaRocheLimitedMemberarvn:CommercialMilestonesMember2026-03-310001655759arvn:VepdegestrantMember2026-01-012026-03-310001655759arvn:VepdegestrantMember2025-01-012025-03-310001655759arvn:PfizerInc.Member2026-01-012026-03-310001655759arvn:PfizerInc.Member2025-01-012025-03-310001655759arvn:RemovalOfCombinationTrialsMember2025-01-012025-03-310001655759arvn:UpdatedResearchTimelinesMember2025-01-012025-03-3100016557592026-04-012026-03-3100016557592027-01-012026-03-3100016557592028-01-012026-03-3100016557592029-01-012026-03-310001655759us-gaap:CorporateBondSecuritiesMemberus-gaap:FairValueInputsLevel2Member2026-03-310001655759us-gaap:USGovernmentDebtSecuritiesMemberus-gaap:FairValueInputsLevel2Member2026-03-310001655759us-gaap:FairValueInputsLevel2Member2026-03-310001655759us-gaap:CorporateBondSecuritiesMemberus-gaap:FairValueInputsLevel2Member2025-12-310001655759us-gaap:USGovernmentDebtSecuritiesMemberus-gaap:FairValueInputsLevel2Member2025-12-310001655759us-gaap:FairValueInputsLevel2Member2025-12-310001655759arvn:LaboratoryEquipmentMember2026-03-310001655759arvn:LaboratoryEquipmentMember2025-12-310001655759us-gaap:LeaseholdImprovementsMember2026-03-310001655759us-gaap:LeaseholdImprovementsMember2025-12-310001655759us-gaap:OfficeEquipmentMember2026-03-310001655759us-gaap:OfficeEquipmentMember2025-12-310001655759srt:MinimumMember2026-03-310001655759stpr:CTarvn:TwoThousandAndEighteenAssistanceAgreementMember2018-09-300001655759stpr:CTarvn:TwoThousandAndEighteenAssistanceAgreementMember2018-09-012018-09-300001655759arvn:EquityDistributionAgreementMemberarvn:PiperSandlerAndCantorMemberarvn:AtTheMarketOfferingMember2023-11-300001655759arvn:EquityDistributionAgreementMemberarvn:PiperSandlerAndCantorMemberarvn:AtTheMarketOfferingMember2026-01-012026-03-310001655759us-gaap:EmployeeStockMember2018-09-300001655759us-gaap:EmployeeStockMember2018-09-012018-09-300001655759us-gaap:EmployeeStockMember2026-03-310001655759us-gaap:EmployeeStockMember2025-01-012025-03-310001655759us-gaap:EmployeeStockMember2026-01-012026-03-310001655759arvn:TwoThousandEighteenStockIncentivePlanMember2018-09-300001655759srt:MaximumMemberarvn:TwoThousandEighteenStockIncentivePlanMember2018-09-300001655759srt:MinimumMemberarvn:TwoThousandEighteenStockIncentivePlanMember2019-01-012019-12-310001655759arvn:TwoThousandEighteenStockIncentivePlanMember2019-01-012019-12-310001655759arvn:TwoThousandEighteenStockIncentivePlanMember2026-03-310001655759arvn:TwoThousandEighteenStockIncentivePlanMemberus-gaap:EmployeeStockMember2026-01-012026-03-310001655759arvn:TwoThousandEighteenStockIncentivePlanMemberus-gaap:EmployeeStockMember2025-01-012025-03-310001655759srt:WeightedAverageMember2026-01-012026-03-310001655759us-gaap:EmployeeStockOptionMember2026-01-012026-03-310001655759us-gaap:EmployeeStockOptionMember2025-01-012025-03-310001655759srt:MinimumMemberus-gaap:EmployeeStockOptionMember2026-01-012026-03-310001655759srt:MaximumMemberus-gaap:EmployeeStockOptionMember2026-01-012026-03-310001655759srt:MinimumMemberus-gaap:EmployeeStockOptionMember2025-01-012025-03-310001655759srt:MaximumMemberus-gaap:EmployeeStockOptionMember2025-01-012025-03-310001655759srt:MinimumMemberus-gaap:EmployeeStockOptionMember2026-03-310001655759srt:MaximumMemberus-gaap:EmployeeStockOptionMember2026-03-310001655759srt:MinimumMemberus-gaap:EmployeeStockOptionMember2025-03-310001655759srt:MaximumMemberus-gaap:EmployeeStockOptionMember2025-03-310001655759arvn:TwoThousandEighteenStockIncentivePlanMember2025-12-310001655759arvn:TwoThousandEighteenStockIncentivePlanMember2025-01-012025-12-310001655759arvn:TwoThousandEighteenStockIncentivePlanMember2026-01-012026-03-310001655759us-gaap:RestrictedStockUnitsRSUMember2026-01-012026-03-310001655759arvn:TwoThousandEighteenStockIncentivePlanMemberus-gaap:RestrictedStockUnitsRSUMember2025-12-310001655759arvn:TwoThousandEighteenStockIncentivePlanMemberus-gaap:RestrictedStockUnitsRSUMember2026-01-012026-03-310001655759arvn:TwoThousandEighteenStockIncentivePlanMemberus-gaap:RestrictedStockUnitsRSUMember2026-03-310001655759us-gaap:RestrictedStockUnitsRSUMember2025-01-012025-03-310001655759us-gaap:EmployeeStockOptionMember2026-01-012026-03-310001655759us-gaap:RestrictedStockUnitsRSUMember2026-01-012026-03-310001655759arvn:YaleUniversityMemberarvn:UpfrontPaymentForAmendedLicenseAgreementAndNovartisLicenseAndAssetAgreementsMember2024-06-012024-06-300001655759arvn:YaleUniversityMemberarvn:MilestonePaymentOnFirstAnniversaryOfSigningMember2024-06-300001655759arvn:MilestonePaymentUponApprovalOfFirstAndSecondRoyaltyProductsMemberarvn:YaleUniversityMembersrt:MaximumMember2024-06-300001655759arvn:YaleUniversityMembersrt:MinimumMember2013-07-052013-07-050001655759arvn:YaleUniversityMembersrt:MinimumMember2013-07-050001655759arvn:LicenseFirstLicensedProductMemberarvn:SuccessBasedMilestonePaymentsMemberarvn:YaleUniversityMember2013-07-052013-07-050001655759arvn:LicenseSecondLicensedProductMemberarvn:SuccessBasedMilestonePaymentsMemberarvn:YaleUniversityMember2013-07-052013-07-050001655759arvn:YaleUniversityMembersrt:MaximumMember2013-07-052013-07-050001655759arvn:ConsultingAgreementLumpSumPaymentMembersrt:DirectorMember2026-03-012026-03-310001655759arvn:ConsultingAgreementCOBRAReimbursementMembersrt:DirectorMember2026-03-012026-03-310001655759srt:DirectorMember2026-03-012026-03-310001655759srt:DirectorMember2026-01-012026-03-3100016557592025-09-012025-09-300001655759us-gaap:ResearchAndDevelopmentExpenseMember2026-01-012026-03-310001655759us-gaap:GeneralAndAdministrativeExpenseMember2026-01-012026-03-310001655759arvn:ReportableSegmentMember2026-01-012026-03-310001655759arvn:ReportableSegmentMember2025-01-012025-03-310001655759arvn:ARV471Memberarvn:ReportableSegmentMember2026-01-012026-03-310001655759arvn:ARV471Memberarvn:ReportableSegmentMember2025-01-012025-03-310001655759arvn:ARV806Memberarvn:ReportableSegmentMember2026-01-012026-03-310001655759arvn:ARV806Memberarvn:ReportableSegmentMember2025-01-012025-03-310001655759arvn:ARV102Memberarvn:ReportableSegmentMember2026-01-012026-03-310001655759arvn:ARV102Memberarvn:ReportableSegmentMember2025-01-012025-03-310001655759arvn:ARV393Memberarvn:ReportableSegmentMember2026-01-012026-03-310001655759arvn:ARV393Memberarvn:ReportableSegmentMember2025-01-012025-03-310001655759arvn:ARV110Memberarvn:ReportableSegmentMember2026-01-012026-03-310001655759arvn:ARV110Memberarvn:ReportableSegmentMember2025-01-012025-03-310001655759arvn:OtherProgramsMemberarvn:ReportableSegmentMember2026-01-012026-03-310001655759arvn:OtherProgramsMemberarvn:ReportableSegmentMember2025-01-012025-03-310001655759arvn:RandyTeelMember2026-01-012026-03-310001655759arvn:AndrewSaikMember2026-01-012026-03-310001655759arvn:NoahBerkowitzMember2026-01-012026-03-310001655759arvn:AngelaCacaceMember2026-01-012026-03-310001655759arvn:DavidLoomisMember2026-01-012026-03-31
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
__________________________________________
FORM 10-Q
__________________________________________
(Mark One)
| | | | | |
| ý | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended March 31, 2026
OR
| | | | | |
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number: 001-38672
__________________________________________
ARVINAS, INC.
(Exact name of registrant as specified in its Charter)
__________________________________________
| | | | | |
| Delaware | 47-2566120 |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
5 Science Park 395 Winchester Ave. New Haven, Connecticut | 06511 |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (203) 535-1456
__________________________________________
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | | | | | | | |
| Title of each class | | Trading Symbol(s) | | Name of each exchange on which registered |
| Common stock, par value $0.001 per share | | ARVN | | The Nasdaq Stock Market LLC |
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ý No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | |
| Large accelerated filer | o | Accelerated filer | ý |
| Non-accelerated filer | o | Smaller reporting company | o |
| | | Emerging growth company | o |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. o
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ý
As of May 7, 2026, the registrant had 64,521,198 shares of common stock, $0.001 par value per share, outstanding.
Table of Contents
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Forward-Looking Statements
This Quarterly Report on Form 10-Q contains forward-looking statements that involve substantial risks and uncertainties. All statements, other than statements of historical facts, contained in this Quarterly Report on Form 10-Q, including statements regarding our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans and objectives of management, are forward-looking statements. The words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “might,” “plan,” “predict,” “project,” “target,” “potential,” “goals,” “will,” “would,” “could,” “should,” “continue” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words.
The forward-looking statements in this Quarterly Report on Form 10-Q include, among other things, statements about:
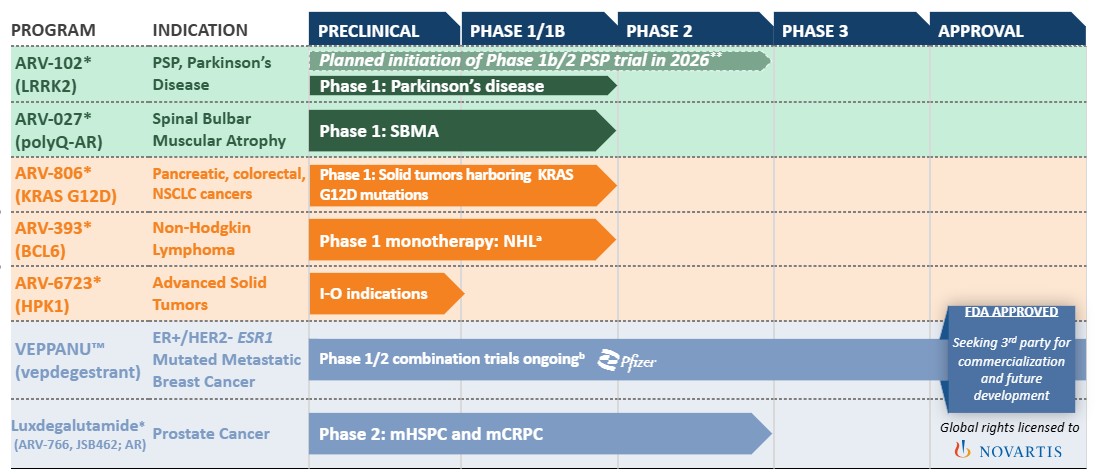
•the initiation, timing, progress and results of our current and/or future clinical trials of ARV-102, ARV-806, ARV-393 and ARV-027, including statements regarding the period during which the results of the clinical trials will become available or the forum in which we will present such results;
•the initiation, timing, progress and results of our current preclinical studies and any future preclinical studies or clinical trials of our other programs, including ARV-6723 and our pan-KRAS degrader, including statements regarding the period during which the results of preclinical studies or clinical trials will become available or the forum in which we will present such results;
•our belief, based on data from our preclinical studies and clinical trials, that PROTAC protein degraders may have distinct advantages over traditional small molecule inhibitors, antibodies and gene-based medicines;
•our belief that PROTAC degraders offer distinct advantages that enable perturbation of protein targets traditionally considered undruggable by conventional therapeutics;
•the timing of, and our ability to obtain, marketing approval of our product candidates and the ability of our product candidates to meet existing or future regulatory standards;
•our plans to pursue research and development of other product candidates;
•the potential advantages of our platform technology and potential advantages and therapeutic benefits of our product candidates;
•our belief that and the extent to which our targeted protein degradation approach may provide distinct advantages over existing therapies and address a broad range of targets, including historically undruggable proteins, in areas of significant unmet need;
•the potential achievement of milestones and receipt of payments under our collaborations, including our collaboration with Pfizer Inc. ("Pfizer") entered into in July 2021;
•our plans, together with Pfizer to jointly select a third party for the commercialization and potential further development of VEPPANU™ (vepdegestrant);
•the potential receipt of payments based on the achievement of milestones related to luxdegalutamide (ARV-766) and future royalties under our license agreement with Novartis Pharma AG;
•the potential payments to be made to Yale University ("Yale") under our amended and restated license agreement with Yale;
•favorable clinical trial results in our ongoing oncology and neurology programs providing further validation of our platform as a new therapeutic modality for the potential treatment of diseases caused by dysregulated intracellular proteins;
•our belief that our leucine-rich repeat kinase 2 ("LRRK2") degraders are particularly well positioned to be evaluated in neurodegenerative diseases where there are currently no disease modifying therapies available, including progressive supranuclear palsy ("PSP") and Parkinson's disease ("PD");
•our belief that the data from our preclinical studies of ARV-102 further support the potential of PROTAC-induced LRRK2 degradation as a treatment for patients with neurodegenerative disease;
•our belief that ARV-806 has the potential to address high unmet need in solid tumors, such as pancreatic, colorectal and non-small cell lung cancer ("NSCLC"), with Kirsten rat sarcoma G12D mutation;
•our expectations with respect to the Phase 1b clinical trial of ARV-102 in the U.S. in patients with PSP and our plans for clinical trials in PSP in the European Union;
•our belief that ARV-806 has the potential to be developed as a monotherapy and in combination with chemotherapy in pancreatic ductal adenocarcinoma and in combination with standard of care ("SOC") treatments in colorectal and non-small cell lung cancer;
•our belief that preclinical data for ARV-806 supports intermittent clinical dosing;
•our belief that PROTAC-mediated degradation has the potential to address the historically undruggable nature of the B-cell lymphoma 6 protein ("BCL6") and that ARV-393 PROTAC-mediated degradation of BCL6 may provide an important novel therapeutic option for patients with non-Hodgkin lymphoma;
•our belief that ARV-393 can be an attractive combination partner for development of novel therapies for lymphoma, including chemo-free combination regimens and/or “all oral” treatment options;
•our belief that the totality of our preclinical data for ARV-393 provides a compelling rationale to evaluate ARV-393 in combination with bi-specifics, oral pathway inhibitors, and potentially other standards of care, in the larger diffuse large B-cell lymphoma indication;
•the potential receipt of revenue from future sales of our product candidates;
•the rate and degree of market acceptance and clinical utility of our product candidates;
•our estimates regarding the potential market opportunity for our product candidates;
•our ability to manage the transition of a new chief executive officer;
•our commercialization plans, and sales, marketing and distribution capabilities and strategy;
•our ability to establish and maintain arrangements for manufacture and testing of our product candidates;
•our ability to enter into additional collaborations with third parties;
•our intellectual property position;
•our plans with respect to our strategy;
•our estimates regarding expenses, future revenues, capital requirements and needs for additional financing, and statements regarding our cash, cash equivalents and marketable securities, including their sufficiency to fund planned operating expenses and capital expenditure requirements into the second half of 2028;
•our belief that non-GAAP financial information, when taken collectively, may be helpful to investors because it provides consistency and comparability with past financial performance;
•the impact of any government laws and regulations; and
•our competitive position.
We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. We have included important factors in the cautionary statements included in our Annual Report on Form 10-K for the year ended December 31, 2025, filed on February 24, 2026, and this Quarterly Report on Form 10-Q, particularly in the “Risk Factors” sections, that we believe could cause actual results or events to differ materially from the forward-looking statements that we make. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments we may make.
You should read this Quarterly Report on Form 10-Q and the documents that we have filed as exhibits to this Quarterly Report on Form 10-Q completely and with the understanding that our actual future results may differ materially from what we expect. We do not assume any obligation to update any forward-looking statements except as required by applicable law.
Throughout this Quarterly Report on Form 10-Q, references to the “Company,” “Arvinas,” “we,” “us,” and “our,” refer to Arvinas, Inc. and its consolidated subsidiaries, except where the context requires otherwise, or any one or more of them as the context may require, and “board of directors” refers to the board of directors of Arvinas, Inc.
The Arvinas name and logo are our trademarks. VEPPANU™ (vepdegestrant) is a trademark of Arvinas Operations, Inc. This Quarterly Report on Form 10-Q contains references to our trademarks and service marks and to those belonging to other entities. Solely for convenience, trademarks and trade names referred to in this Quarterly Report on Form 10-Q, including logos, artwork and other visual displays, may appear without the ® or ™ symbols, but such references are not intended to indicate in any way that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other entities’ trade names, trademarks or service marks to imply a relationship with, or endorsement or sponsorship of us by, any other entity.
PART I—FINANCIAL INFORMATION
Item 1. Financial Statements.
ARVINAS, INC. AND SUBSIDIARIES
Condensed Consolidated Balance Sheets (unaudited)
| | | | | | | | | | | |
| (dollars and shares in millions, except per share amounts) | March 31,
2026 | | December 31,
2025 |
| Assets | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 87.3 | | | $ | 142.9 | |
| | | |
| Marketable securities | 527.6 | | | 542.5 | |
| Accounts receivable | 1.8 | | | 1.0 | |
| Other receivables | 4.6 | | | 5.4 | |
| Prepaid expenses and other current assets | 9.4 | | | 8.9 | |
| Total current assets | 630.7 | | | 700.7 | |
| Property, equipment and leasehold improvements, net | 5.5 | | | 5.2 | |
| Operating lease right-of-use assets | 7.8 | | | 8.2 | |
| Collaboration contract asset and other assets | 3.5 | | | 3.8 | |
| Total assets | $ | 647.5 | | | $ | 717.9 | |
| Liabilities and stockholders' equity | | | |
| Current liabilities: | | | |
| Accounts payable and accrued liabilities | $ | 62.3 | | | $ | 69.5 | |
| Deferred revenue | 51.8 | | | 71.3 | |
| | | |
| Current portion of operating lease liabilities | 1.8 | | | 1.7 | |
| Total current liabilities | 115.9 | | | 142.5 | |
| Deferred revenue | 138.2 | | | 134.3 | |
| Long-term debt | 0.3 | | | 0.4 | |
| Operating lease liabilities | 6.3 | | | 6.8 | |
| Total liabilities | 260.7 | | | 284.0 | |
| Commitments and Contingencies (Note 12) | | | |
| Stockholders’ equity: | | | |
Preferred stock, $0.001 par value, zero shares issued and outstanding as of March 31, 2026 and December 31, 2025, respectively | — | | | — | |
Common stock, $0.001 par value; 74.5 shares issued and 64.5 shares outstanding as of March 31, 2026, and 73.5 shares issued and 63.5 outstanding as of December 31, 2025 | 0.1 | | | 0.1 | |
| Accumulated deficit | (1,670.0) | | | (1,612.4) | |
| Additional paid-in capital | 2,149.0 | | | 2,136.9 | |
| Accumulated other comprehensive (loss) income | (0.4) | | | 1.2 | |
Treasury Stock, at cost (10.0 shares as of March 31, 2026 and December 31, 2025) | (91.9) | | | (91.9) | |
| Total stockholders’ equity | 386.8 | | | 433.9 | |
| Total liabilities and stockholders’ equity | $ | 647.5 | | | $ | 717.9 | |
See accompanying notes to the condensed consolidated financial statements
ARVINAS, INC. AND SUBSIDIARIES
Condensed Consolidated Statements of Operations and Comprehensive (Loss) Income (unaudited)
| | | | | | | | | | | | | | | |
| (dollars and shares in millions, except per share amounts) | For the Three Months Ended
March 31, | | |
| Consolidated Statements of Operations | 2026 | | 2025 | | | | |
| Revenue | $ | 15.6 | | | $ | 188.8 | | | | | |
| Operating expenses: | | | | | | | |
| Research and development | 60.3 | | | 90.8 | | | | | |
| General and administrative | 19.1 | | | 26.6 | | | | | |
| Total operating expenses | 79.4 | | | 117.4 | | | | | |
| (Loss) income from operations | (63.8) | | | 71.4 | | | | | |
| Other income | | | | | | | |
| Other expense, net | (0.1) | | | — | | | | | |
| Interest income, net | 6.4 | | | 11.7 | | | | | |
| | | | | | | |
| Total other income | 6.3 | | | 11.7 | | | | | |
| Net (loss) income before income taxes | (57.5) | | | 83.1 | | | | | |
| Income tax expense | (0.1) | | | (0.2) | | | | | |
| | | | | | | |
| Net (loss) income | $ | (57.6) | | | $ | 82.9 | | | | | |
| | | | | | | |
| (Loss) earnings per common share | | | | | | | |
| Basic | $ | (0.90) | | | $ | 1.14 | | | | | |
| Diluted | $ | (0.90) | | | $ | 1.14 | | | | | |
| | | | | | | |
Weighted average common shares outstanding | | | | | | | |
| Basic | 64.0 | | | 72.5 | | | | | |
| Diluted | 64.0 | | | 72.7 | | | | | |
| | | | | | | | | | | | | | | |
| (dollars in millions) | For the Three Months Ended
March 31, | | |
| Consolidated Statements of Comprehensive (Loss) Income | 2026 | | 2025 | | | | |
| Net (loss) income | $ | (57.6) | | | $ | 82.9 | | | | | |
| Other comprehensive loss: | | | | | | | |
| Unrealized (loss) gain on available-for-sale securities | (1.6) | | | 0.5 | | | | | |
| Comprehensive (loss) income | $ | (59.2) | | | $ | 83.4 | | | | | |
See accompanying notes to the condensed consolidated financial statements
ARVINAS, INC. AND SUBSIDIARIES
Condensed Consolidated Statements of Changes in Stockholders’ Equity (unaudited)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| (dollars and shares in millions) | Common | | Accumulated
Deficit | | Additional
Paid-in
Capital | | Accumulated
Other
Comprehensive
Income (Loss) | | Treasury | | Total
Stockholders'
Equity |
For the Three Months Ended March 31, 2026 and 2025 | Shares | | Amount | | | | | Shares | | Amount | |
| Balance as of December 31, 2025 | 73.5 | | | $ | 0.1 | | | $ | (1,612.4) | | | $ | 2,136.9 | | | $ | 1.2 | | | 10.0 | | | $ | (91.9) | | | $ | 433.9 | |
| Stock-based compensation | — | | | — | | | — | | | 12.1 | | | — | | | — | | | — | | | 12.1 | |
| Net loss | — | | | — | | | (57.6) | | | — | | | — | | | — | | | — | | | (57.6) | |
| | | | | | | | | | | | | | | |
| Issuance of common stock under equity incentive plans | 1.0 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Unrealized loss on available-for-sale securities | — | | | — | | | — | | | — | | | (1.6) | | | — | | | — | | | (1.6) | |
Balance as of March 31, 2026 | 74.5 | | | 0.1 | | | (1,670.0) | | | 2,149.0 | | | (0.4) | | | 10.0 | | | (91.9) | | | 386.8 | |
| Balance as of December 31, 2024 | 68.8 | | | $ | 0.1 | | | $ | (1,531.6) | | | $ | 2,092.2 | | | $ | 1.0 | | | — | | | $ | — | | | $ | 561.7 | |
| Stock-based compensation | — | | | — | | | — | | | 15.0 | | | — | | | — | | | — | | | 15.0 | |
| Net income | — | | | — | | | 82.9 | | | — | | | — | | | — | | | — | | | 82.9 | |
| | | | | | | | | | | | | | | |
| Issuance of common stock under equity incentive plans | 0.8 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Issuance of common stock for pre-funded warrants | 3.4 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| | | | | | | | | | | | | | | |
| Unrealized gain on available-for-sale securities | — | | | — | | | — | | | — | | | 0.5 | | | — | | | — | | | 0.5 | |
Balance as of March 31, 2025 | 73.0 | | | $ | 0.1 | | | $ | (1,448.7) | | | $ | 2,107.2 | | | $ | 1.5 | | | — | | | $ | — | | | $ | 660.1 | |
See accompanying notes to the condensed consolidated financial statements
ARVINAS, INC. AND SUBSIDIARIES
Condensed Consolidated Statements of Cash Flows (unaudited)
| | | | | | | | | | | |
| For the Three Months Ended
March 31, |
| (dollars in millions) | 2026 | | 2025 |
| Cash flows from operating activities: | | | |
| Net (loss) income | $ | (57.6) | | | $ | 82.9 | |
| Adjustments to reconcile net (loss) income to net cash used in operating activities: | | | |
| Depreciation and amortization | 0.7 | | | 0.7 | |
| Net accretion of bond discounts/premiums | (1.7) | | | (3.8) | |
| | | |
| | | |
| Amortization of right-of-use assets | 0.4 | | | 0.6 | |
| Amortization of collaboration contract asset | 0.3 | | | 3.3 | |
| | | |
| Stock-based compensation | 12.1 | | | 15.0 | |
| Changes in operating assets and liabilities: | | | |
| Accounts receivable | (0.8) | | | 5.3 | |
| Other receivables | 0.8 | | | — | |
| Prepaid expenses and other assets | (0.6) | | | (2.7) | |
| | | |
| Accounts payable and accrued liabilities | (6.8) | | | (0.8) | |
| Operating lease liability | (0.4) | | | (0.5) | |
| Deferred revenue | (15.6) | | | (188.9) | |
| Net cash used in operating activities | (69.2) | | | (88.9) | |
| Cash flows from investing activities: | | | |
| Purchases of marketable securities | (178.9) | | | (119.6) | |
| Maturities of marketable securities | 165.0 | | | 189.5 | |
| Sales of marketable securities | 28.9 | | | — | |
| Purchases of property, equipment and leasehold improvements | (1.3) | | | (0.4) | |
| | | |
| Net cash provided by investing activities | 13.7 | | | 69.5 | |
| Cash flows from financing activities: | | | |
| Repayments of long-term debt | (0.1) | | | (0.1) | |
| | | |
| | | |
| | | |
| | | |
| Net cash used in financing activities | (0.1) | | | (0.1) | |
| Net decrease in cash and cash equivalents | (55.6) | | | (19.5) | |
| Cash and cash equivalents, beginning of the period | 142.9 | | | 100.5 | |
| Cash and cash equivalents, end of the period | $ | 87.3 | | | $ | 81.0 | |
| Supplemental disclosure of cash flow information: | | | |
| Purchases of property, equipment and leasehold improvements unpaid at period end | $ | — | | | $ | 0.1 | |
| | | |
| | | |
| | | |
| | | |
See accompanying notes to the condensed consolidated financial statements
ARVINAS, INC. AND SUBSIDIARIES
Notes to Condensed Consolidated Financial Statements (unaudited)
1. Nature of Business and Basis of Presentation
Arvinas, Inc. and its subsidiaries (“Arvinas” or the "Company”) is a biotechnology company dedicated to improving the lives of patients suffering from debilitating and life-threatening diseases.
The accompanying unaudited condensed consolidated financial statements include the accounts of Arvinas, Inc. and its subsidiaries. The financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) for interim financial information and the instructions to Form 10-Q and Article 10 of Regulation S-X under the Securities Exchange Act of 1934, as amended (“Exchange Act”). Certain information and footnote disclosures normally included in annual financial statements prepared in accordance with U.S. GAAP have been condensed or omitted pursuant to U.S. Securities and Exchange Commission (“SEC”) rules. In the opinion of management, all adjustments (consisting of normal recurring adjustments) necessary for a fair presentation have been included. The condensed consolidated balance sheet as of December 31, 2025 has been derived from the Company's audited consolidated financial statements as of that date. The financial statements should be read in conjunction with the audited consolidated financial statements and notes thereto for the year ended December 31, 2025, forming part of Arvinas’ 2025 Annual Report on Form 10-K filed with the SEC on February 24, 2026.
The preparation of the Company’s unaudited condensed consolidated financial statements in conformity with U.S. GAAP requires management to make certain estimates and assumptions that affect the reported amount of assets, liabilities, revenue and expenses. These estimates include assumptions and judgments based on historical experience, current conditions, future expectations and other factors the Company considers reasonable. These estimates are reviewed on an ongoing basis and revised as necessary. Actual results could differ from these estimates.
Risks and Uncertainties
The Company is subject to a number of risks similar to other biotechnology companies in a similar stage, including, but not limited to, the need to obtain adequate additional funding, possible failure of preclinical testing or clinical trials, the need to obtain marketing approval for its product candidates, competitors developing new technological innovations, and the need to successfully commercialize and gain market acceptance of the Company’s products and to protect its proprietary technology. If the Company does not successfully obtain regulatory approval of its product candidates, it will be unable to generate revenue from product sales or achieve profitability.
To date, the Company has not generated any revenue from product sales and expects to incur additional operating losses and negative operating cash flows for the foreseeable future. The Company has financed its operations primarily through sales of assets and equity interests, proceeds from collaborations and a licensing arrangement, grant funding and debt financing. The Company had cash, cash equivalents and marketable securities of approximately $614.9 million as of March 31, 2026.
2. Summary of Accounting Pronouncements and Significant Accounting Policies
Accounting Pronouncements
Recently Adopted Accounting Pronouncements
There have been no recently adopted accounting pronouncements that have had a material impact on the Company's unaudited condensed consolidated financial statements.
Recently Issued Accounting Pronouncements Not Yet Adopted
Income Statement - Reporting Comprehensive Income - Expense Disaggregation Disclosures (Subtopic 220-40) - In November 2024, the Financial Accounting Standards Board (“FASB”) issued Accounting
Standards Update (“ASU”) No. 2024-03, "Disaggregation of Income Statement Expenses," which requires disclosures of certain disaggregated income statement expense captions into specified categories within the footnotes to the financial statements. The requirements of the ASU are effective for annual periods beginning after December 15, 2026 and interim reporting periods beginning after December 15, 2027, with early adoption permitted. The requirements will be applied prospectively with the option for retrospective application. The Company is currently evaluating the impact ASU No. 2024-03 will have on its condensed consolidated financial statements.
Significant Accounting Policies
There were no changes to the Company’s significant accounting policies during the three months ended March 31, 2026.
3. Research Collaboration and License Agreements
Vepdegestrant (ARV-471) Collaboration Agreement
In July 2021, the Company entered into a Collaboration Agreement with Pfizer Inc. (“Pfizer”) (the “Vepdegestrant (ARV-471) Collaboration Agreement”) pursuant to which the Company granted Pfizer worldwide co-exclusive rights to develop and commercialize products containing the Company’s proprietary compound vepdegestrant (the “Licensed Products”). Under the Vepdegestrant (ARV-471) Collaboration Agreement, the Company received an upfront, non-refundable payment of $650.0 million. In addition, the Company is eligible to receive up to an additional $1.4 billion in contingent payments based on specific regulatory and sales-based milestones for the Licensed Products. Of the total contingent payments, $400.0 million in regulatory milestones are related to marketing approvals and $1.0 billion are related to sales-based milestones. There were no regulatory or sales-based milestone payments received through March 31, 2026.
The Company and Pfizer share equally all development costs for the Licensed Products, including costs of conducting clinical trials, subject to certain exceptions.
The Company and Pfizer share equally all development costs for the Licensed Products, including costs of conducting clinical trials, subject to certain exceptions. Except for certain regions described below, the parties will also share equally all profits and losses in commercialization and medical affairs activities for the Licensed Products in all other countries, subject to certain exceptions.
As a direct result of the Company’s entry into the Vepdegestrant (ARV-471) Collaboration Agreement, the Company incurred direct and incremental costs to obtain the contract, paid to a financial advisor, totaling $12.9 million. In accordance with Accounting Standards Codification ("ASC") 340, Other Assets and Deferred Costs, the Company recognized an asset of $12.9 million in collaboration contract asset and other assets in the condensed consolidated balance sheet at inception of the Vepdegestrant (ARV-471) Collaboration Agreement, which is being amortized as general and administrative expense over the total estimated period of performance under the Vepdegestrant (ARV-471) Collaboration Agreement.
In the second quarter of 2026, the Company announced that the U.S. Food and Drug Administration ("FDA") has granted approval for VEPPANU™ (vepdegestrant) for the treatment of adults with estrogen receptor-positive (“ER+”)/human epidermal growth factor receptor 2-negative (“HER2-”), estrogen receptor 1 (“ESR1”)-mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endrocrine-based therapy.
Pursuant to the Vepdegestrant (ARV-471) Collaboration Agreement, the Company will receive $50.0 million as a development milestone payment in connection with the FDA’s approval of VEPPANU (the “Milestone Payment”). The Milestone Payment will be offset by certain amounts that the Company will owe to Yale University ("Yale") pursuant to the amended and restated license agreement, dated June 18, 2024, by and between the Company, one of its subsidiaries, and Yale (the "Yale Agreement").
In September 2025, the Company announced that the Company and Pfizer have agreed to jointly select a third party for the commercialization and potential further development of vepdegestrant. The Company and its collaborator, Pfizer Inc., remain on track to announce selection of a third party to commercialize VEPPANU.
Pfizer Research Collaboration Agreement
In December 2017, the Company entered into a Research Collaboration and License Agreement with Pfizer (the “Pfizer Research Collaboration Agreement”). Under the terms of the Pfizer Research Collaboration Agreement, the Company received an upfront, non-refundable payment and certain additional payments totaling $28.0 million in 2018 in exchange for use of the Company’s technology license and to fund Pfizer-related research as defined within the Pfizer Research Collaboration Agreement. These payments are being recognized over the total estimated period of performance. As of March 31, 2026, there remains a single target under the Pfizer Research Collaboration Agreement, and, in accordance with the terms of such Agreement, the Company is eligible to receive up to an additional $3.8 million in non-refundable option payments if Pfizer exercises its option for such target protein under the Pfizer Research Collaboration Agreement.
The Company is also entitled to receive up to $225.0 million in development milestone payments and up to $550.0 million in sales-based milestone payments for all designated target proteins under the Pfizer Research Collaboration Agreement, as well as tiered royalties based on sales, which may be subject to reductions. There were no sales-based milestone payments or royalties received through March 31, 2026.
Novartis License and Asset Agreements
In April 2024, the Company entered into a transaction (the "Novartis Transaction"), including both a license agreement (the "Novartis License Agreement") and an asset purchase agreement (the "Novartis Asset Agreement") with Novartis Pharma AG ("Novartis") for the worldwide development, manufacture and commercialization of luxdegalutamide (ARV-766), the Company's second generation PROTAC androgen receptor (AR) degrader for patients with prostate cancer and for the sale of the Company's preclinical AR-V7 program. Under the terms of the agreements, Novartis is responsible for worldwide clinical development and commercialization of luxdegalutamide (ARV-766) and has all research, development, manufacturing, and commercialization rights with respect to the Company’s PROTAC protein degrader targeting AR-V7, a splice variant of the AR.
In May 2024, Novartis paid to the Company a one-time, upfront payment in the aggregate amount of $150.0 million in accordance with the terms of the Novartis License Agreement and the Novartis Asset Agreement. The upfront payment was recognized as revenue over the performance period, which concluded as of December 31, 2024 as the technology transfer period ended as the Company completed the transition of its ongoing and planned clinical trials of luxdegalutamide (ARV-766) to Novartis.
Under the terms of the Novartis License Agreement, the Company is eligible to receive up to an additional $1.01 billion as contingent payments based on specified development, regulatory and commercial milestones for luxdegalutamide (ARV-766) being met, as well as tiered royalties based on worldwide net sales of luxdegalutamide (ARV-766), subject to reduction under certain circumstances as provided in the Novartis License Agreement. There were no development, regulatory or commercial milestone payments, or sales-based royalties received during the three months ended March 31, 2026 and 2025.
The Novartis License Agreement will continue on a country-by-country basis (or, in certain cases, a region-by-region basis) until the expiration of the applicable royalty term for such country (or region, as applicable). The Novartis License Agreement contains customary termination provisions, including that either party may terminate the Novartis License Agreement (a) upon the material breach of the other party or (b) in the event the other party experiences an insolvency event. Additionally, Novartis may terminate the Novartis License Agreement for convenience or upon a safety or regulatory issue.
Restated Genentech Agreement
In November 2017, the Company entered into an Amended and Restated Option, License, and Collaboration Agreement (the “Restated Genentech Agreement”) with Genentech, Inc. and F. Hoffman-La Roche Ltd. (together "Genentech"), amending a previous Genentech agreement entered into in September 2015. Under the Restated Genentech Agreement, the Company received additional upfront, non-refundable payments of $34.5 million (in addition to $11.0 million received under the previous agreement in 2015) to fund Genentech-related research. Upfront non-refundable payments were recognized as revenue over the performance period, which concluded during the first quarter of 2023. The research phase of the collaboration with Genentech has ended. As such, Genentech is no longer able to nominate new targets into the
collaboration. The only target that remains part of the collaboration is the PROTAC targeted protein degrader for which Genentech exercised its exclusive option upon amendment and restatement of the agreement.
The Company is eligible to receive up to $44.0 million per target protein in development milestone payments, $52.5 million in regulatory milestone payments and $60.0 million in commercial milestone payments based on sales as well as tiered royalties based on sales. There were no development, regulatory or commercial milestone payments or royalties received through March 31, 2026.
During the three months ended March 31, 2026 and 2025, the Company's sources of revenue were as follows:
| | | | | | | | | | | |
| (dollars in millions) | March 31,
2026 | | March 31,
2025 |
| Revenue | | | |
| Vepdegestrant (ARV-471) Collaboration Agreement | $ | 14.4 | | | $ | 189.9 | |
| Pfizer Research Collaboration Agreement | 1.2 | | | (1.1) | |
| Total Revenue | $ | 15.6 | | | $ | 188.8 | |
| | | |
During the three months ended March 31, 2025, the Company updated its estimate to satisfy the performance obligations under the Vepdegestrant (ARV-471) Collaboration Agreement due to the removal of the first-line Phase 3 combination trial with Pfizer’s novel investigational CDK4 inhibitor, atirmociclib, and the removal of the second-line Phase 3 combination trial with a CDK4/6 inhibitor from the development plan. The change in accounting estimate resulted in an increase in revenue of $150.2 million, an increase in operating expenses of $2.6 million, a decrease in net loss of $147.6 million, and an increase in basic and diluted earnings per share of $2.04 and $2.03, respectively, for the three months ended March 31, 2025.
During the three months ended March 31, 2025, the Company also changed its estimate of the duration of the performance period under the Pfizer Research Collaboration Agreement as a result of updated research timelines. The change in accounting estimate resulted in a decrease in revenue and net income of $2.5 million , and a decrease in basic and diluted loss per share of $0.03 for the three months ended March 31, 2025. The reversed revenue will continue to be recognized in future periods as the Company continues to advance on the performance obligation under the updated collaboration timeline.
During the three months ended March 31, 2026, no changes in accounting estimates related to the Company's collaborations were recorded.
Changes in the Company's contract balances for the three months ended March 31, 2026 and 2025 were as follows:
| | | | | | | | | | | |
| (dollars in millions) | March 31,
2026 | | March 31,
2025 |
| Accounts receivable related to collaborations | | | |
| Beginning balance | $ | 1.0 | | | $ | 5.7 | |
| Additions | 0.8 | | | — | |
| Payments received | — | | | (5.3) | |
| Ending balance | $ | 1.8 | | | $ | 0.4 | |
| Accounts payable related to collaborations | | | |
| Beginning balance | $ | 16.8 | | | $ | 5.4 | |
| Additions | 9.1 | | | 19.4 | |
| Payments made | — | | | (5.4) | |
| Ending balance | $ | 25.9 | | | $ | 19.4 | |
| Contract assets: Collaboration contract asset | | | |
| Beginning balance | $ | 3.5 | | | $ | 7.8 | |
| | | |
| Amortization | (0.3) | | | (3.3) | |
| Ending balance | $ | 3.2 | | | $ | 4.5 | |
| Contract liabilities: Deferred revenue | | | |
| Beginning balance | $ | 205.6 | | | $ | 448.2 | |
| | | |
| Revenue recognized from balances held at the beginning of the period | (15.6) | | | (188.8) | |
| | | |
| Ending balance | $ | 190.0 | | | $ | 259.4 | |
The aggregate amount of the transaction price allocated to performance obligations that were unsatisfied as of March 31, 2026 totaled $190.0 million, which is expected to be recognized in the following periods:
| | | | | |
| (dollars in millions) | |
| Remainder of 2026 | $ | 46.7 | |
| 2027 | 20.3 | |
| 2028 | 20.3 | |
| 2029 | 102.7 | |
| |
| |
| |
| Total | $ | 190.0 | |
4. Marketable Securities and Fair Value Measurements
The following is a summary of the Company’s available-for-sale marketable securities measured at fair value on a recurring basis.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | March 31, 2026 |
| (dollars in millions) | | Valuation Hierarchy | | Amortized Cost | | Gross Unrealized Gains | | Gross Unrealized Losses | | Fair Value |
| Corporate bonds | | Level 2 | | $ | 503.6 | | | $ | 0.3 | | | $ | (0.7) | | | $ | 503.2 | |
| Government securities | | Level 2 | | 24.4 | | | — | | | — | | | 24.4 | |
| Total | | | | $ | 528.0 | | | $ | 0.3 | | | $ | (0.7) | | | $ | 527.6 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | December 31, 2025 |
| (dollars in millions) | | Valuation Hierarchy | | Amortized Cost | | Gross Unrealized Gains | | Gross Unrealized Losses | | Fair Value |
| Corporate bonds | | Level 2 | | $ | 528.0 | | | $ | 1.1 | | | $ | — | | | $ | 529.1 | |
| Government securities | | Level 2 | | 13.3 | | | 0.1 | | | — | | | 13.4 | |
| Total | | | | $ | 541.3 | | | $ | 1.2 | | | $ | — | | | $ | 542.5 | |
The Company generally does not intend to sell any investments prior to recovery of their amortized cost basis for any investment in an unrealized loss position. As such, the Company has classified these losses as temporary in nature.
The carrying value of cash and cash equivalents, accounts receivable and accounts payable and accrued liabilities approximate their fair values due to the short-term nature of these assets and liabilities.
5. Property, Equipment and Leasehold Improvements
Property, equipment and leasehold improvements consist of the following:
| | | | | | | | | | | |
| (dollars in millions) | March 31,
2026 | | December 31,
2025 |
| Laboratory equipment | $ | 21.7 | | | $ | 21.3 | |
| Leasehold improvements | 9.1 | | | 9.1 | |
| Office equipment | 3.2 | | | 3.0 | |
| Total property, equipment and leasehold improvements | 34.0 | | | 33.4 | |
| Less: accumulated depreciation and amortization | (28.5) | | | (28.2) | |
| Property, equipment and leasehold improvements, net | $ | 5.5 | | | $ | 5.2 | |
During each of the three months ended March 31, 2026 and 2025, the Company recognized depreciation and amortization expense of $0.7 million.
6. Right-of-Use Assets and Liabilities
Operating lease liabilities and their corresponding right-of-use ("ROU") assets are recorded based on the present value of lease payments over the expected remaining lease term. The interest rate implicit in lease contracts is typically not readily determinable. As a result, the Company utilizes its incremental borrowing rate, which reflects the fixed rate at which it could borrow on a collateralized basis the amount of the lease payments in the same currency, for a similar term, in a similar economic environment. The Company's weighted average incremental borrowing rate at March 31, 2026 totaled 7.0%. Lease expense is recognized on a straight-line basis over the lease term.
The Company has an operating lease, as amended, for its corporate office and laboratories, which expires no later than December 2029. The lease has a weighted average remaining term of approximately 3.8 years.
The components of lease expense were as follows:
| | | | | | | | | | | | | | | |
| | Three Months Ended
March 31, | | |
| (dollars in millions) | 2026 | | 2025 | | | | |
| Operating lease cost | $ | 0.6 | | | $ | 0.7 | | | | | |
Supplemental cash flow information related to leases was as follows:
| | | | | | | | | | | |
| Three Months Ended
March 31, |
| (dollars in millions) | 2026 | | 2025 |
| Cash paid for amounts included in the measurement of lease liabilities: | | | |
| Operating cash flows from operating leases | $ | 0.4 | | | $ | 0.5 | |
| Supplemental non-cash information: | | | |
| Right-of-use assets obtained in exchange for new lease obligations | $ | — | | | $ | 1.5 | |
Maturities of operating lease liabilities as of March 31, 2026, were as follows:
| | | | | |
| (dollars in millions) | |
| Remainder of 2026 | $ | 1.7 | |
| 2027 | 2.4 | |
| 2028 | 2.5 | |
| 2029 | 2.6 | |
| 2030 | — | |
| |
| |
| Total lease payments | 9.2 | |
| Less: imputed interest | (1.1) | |
| Total | $ | 8.1 | |
7. Accounts Payable and Accrued Liabilities
Accounts payable and accrued liabilities consisted of the following:
| | | | | | | | | | | |
| (dollars in millions) | March 31,
2026 | | December 31,
2025 |
| Accounts payable | $ | 31.4 | | | $ | 24.4 | |
| Accrued liabilities | | | |
| Research and development expenses | 17.4 | | | 18.2 | |
| Employee expenses | 4.5 | | | 17.5 | |
| Income taxes | 5.0 | | | 4.8 | |
| General and administrative and commercial expenses | 2.4 | | | 3.7 | |
| Professional fees | 1.6 | | | 0.9 | |
| Total accounts payable and accrued liabilities | $ | 62.3 | | | $ | 69.5 | |
8. Long-Term Debt
Debt obligations consisted of the following:
| | | | | | | | | | | | | | | | | | | | | | | |
| (dollars in millions) | Maturity Date | | Interest Rate | | March 31,
2026 | | December 31,
2025 |
2018 Assistance Agreement Debt | 09/28 | | 3.25% | | $ | 0.5 | | | $ | 0.6 | |
| Less: current installments included within Accounts payable and accrued liabilities | | | | | (0.2) | | | (0.2) | |
| Total long-term debt | | | | | $ | 0.3 | | | $ | 0.4 | |
In June 2018, the Company entered into an assistance agreement with the State of Connecticut (the "2018 Assistance Agreement") to provide funding for the expansion and renovation of laboratory and office space. The Company borrowed $2.0 million under the 2018 Assistance Agreement in September 2018, of which
$1.0 million was forgiven upon meeting certain employment conditions. Borrowings under the 2018 Assistance Agreement bear an interest rate of 3.25% per annum, with interest-only payments required for the first 60 months, and mature in September 2028. The 2018 Assistance Agreement requires that the Company be located in the State of Connecticut through September 2028, with a default penalty of repayment of the full original funding amount of $2.0 million plus liquidated damages of 7.5% of the total amount of funding received.
Minimum future principal payments on long-term debt as of March 31, 2026 are as follows:
| | | | | |
| (dollars in millions) | |
| Remainder of 2026 | $ | 0.1 | |
| 2027 | 0.2 | |
| 2028 | 0.2 | |
| |
| |
| Total | $ | 0.5 | |
During the three months ended March 31, 2026 and 2025, interest expense was immaterial.
9. Equity
Equity Distribution Agreements
In November 2023, the Company amended and restated the Equity Distribution Agreement with Piper Sandler & Company (“Piper Sandler”) and Cantor Fitzgerald & Co. (“Cantor”), as agents, pursuant to which the Company may offer and sell from time to time, through the agents, up to approximately $262.8 million of the common stock registered under a universal shelf registration statement pursuant to one or more “at-the-market” offerings. During the three months ended March 31, 2026, no shares were issued under this agreement.
Stock-based Compensation
2018 Employee Stock Purchase Plan
In September 2018, the Company adopted the 2018 Employee Stock Purchase Plan (the “2018 ESPP”), with the first offering period under the 2018 ESPP commencing on January 1, 2020, by initially providing participating employees with the opportunity to purchase an aggregate of 311,850 shares of the Company’s common stock. The number of shares of the Company’s common stock reserved for issuance under the 2018 ESPP increased, pursuant to the terms of the 2018 ESPP, by additional shares equal to 1% of the Company’s then-outstanding common stock, effective as of January 1 of each year. As of March 31, 2026, 4,212,347 shares remained available for purchase. During the three months ended March 31, 2026 and 2025, no shares of common stock were issued under the 2018 ESPP.
2018 Stock Incentive Plan
In September 2018, the Company’s board of directors adopted, and the Company’s stockholders approved, the 2018 Stock Incentive Plan (the “2018 Plan”), which became effective upon the effectiveness of the registration statement on Form S-1 for the Company’s initial public offering. The number of shares of common stock initially available for issuance under the 2018 Plan equaled the sum of (1) 4,067,007 shares of common stock; plus (2) the number of shares of common stock (up to 1,277,181 shares) issued in respect of incentive units granted under the Fourth Amendment to the Company’s Incentive Share Plan, which was terminated in September 2018, that were subject to vesting immediately prior to the effectiveness of the registration statement that expire, terminate or are otherwise surrendered, canceled, forfeited or repurchased by the Company at their original issuance price pursuant to a contractual repurchase right; plus (3) an annual increase on the first day of each fiscal year beginning with the fiscal year ended December 31, 2019 and continuing to, and including, the fiscal year ending December 31, 2028, equal to the lesser of 4,989,593 shares of the Company’s common stock, 4% of the number of shares of the Company’s common stock outstanding on the first day of the year or an amount determined by the Company’s board of directors. As of March 31, 2026, 2,981,638 shares remained available for issuance under the 2018 Plan. Shares of common stock subject to outstanding equity awards that expire or are terminated, surrendered or canceled without having been fully exercised or are forfeited in whole or in part are available for future grants of awards.
Compensation Expense
During the three months ended March 31, 2026 and 2025, the Company recognized compensation expense of $12.1 million and $15.0 million, respectively, related to the issuance of incentive awards, including $0.1 million and $0.2 million, respectively, related to the 2018 ESPP.
As of March 31, 2026, there was $45.8 million of total unrecognized compensation expense that is expected to be amortized over a weighted average period of approximately 1.9 years.
Stock Options
The fair value of the stock options granted during the three months ended March 31, 2026 and 2025 was determined using the Black-Scholes option pricing model with the following assumptions:
| | | | | | | | | | | |
| | March 31,
2026 | | March 31,
2025 |
Expected volatility (1) | 77.5% - 77.5% | | 72.1% - 72.2% |
Expected term (years) (2) | 5.6 - 5.6 | | 5.5 - 5.6 |
Risk free interest rate (3) | 3.6% - 3.6% | | 4.0% - 4.4% |
| Expected dividend yield | 0 | % | | 0 | % |
| Exercise price | $13.38 - $13.38 | | $16.23 - $17.70 |
(1) Expected volatility is calculated by utilizing the Company's historical volatility of its stock price over a period equal to the expected term.
(2) Expected term is calculated based on the Company's historical experience.
(3) Risk free interest rate is based on an interpolation of U.S. Treasury rates to reflect the expected term at the date of grant.
A summary of the stock option activity under the 2018 Plan during the three months ended March 31, 2026 is presented below. Included in the table are stock options granted to employees, directors and consultants under the 2018 Plan, as well as options to purchase 255,611 shares of common stock granted to certain employees pursuant to the Nasdaq inducement grant exception in accordance with Nasdaq Listing Rule 5635(c)(4).
| | | | | | | | | | | | | | | | | | | | | | | | | | |
(dollars in millions,
except weighted average exercise price) | | Options | | Weighted Average Exercise Price | | Weighted Average Remaining Contractual Term (Years) | | Aggregate Intrinsic Value |
Outstanding as of December 31, 2025 | | 8,631,075 | | | $ | 35.89 | | | 6.5 | | $ | 5.3 | |
| Granted | | 905,449 | | | $ | 13.38 | | | | | |
| | | | | | | | |
| Cancelled / Forfeited | | (144,146) | | | $ | 45.57 | | | | | |
| | | | | | | | |
Outstanding as of March 31, 2026 | | 9,392,378 | | | $ | 33.57 | | | 6.6 | | $ | 3.9 | |
Vested and exercisable as of March 31, 2026 | | 6,202,436 | | | $ | 43.09 | | | 5.3 | | $ | — | |
Vested and expected to vest as of March 31, 2026 | | 9,000,947 | | | $ | 34.35 | | | 6.5 | | $ | 3.7 | |
The weighted-average grant date fair value per share of options granted during the three months ended March 31, 2026 and 2025 was $9.06 and $11.50, respectively. There were no options exercised during the three months ended March 31, 2026 and 2025.
Restricted Stock Units ("RSUs")
A summary of RSU activity under the 2018 Plan during the three months ended March 31, 2026 is presented below. Included in the table are RSUs granted to employees, directors and consultants under the
2018 Plan, as well as RSUs representing 127,774 shares of common stock granted to certain employees pursuant to the Nasdaq inducement grant exception in accordance with Nasdaq Listing Rule 5635(c)(4).
| | | | | | | | | | | | | | |
| | Shares | | Weighted Average Grant Date Fair Value Per Share |
Unvested RSUs as of December 31, 2025 | | 3,624,051 | | | $ | 18.59 | |
| Granted | | 1,853,715 | | | $ | 13.38 | |
| Vested | | (979,308) | | | $ | 31.80 | |
| Cancelled / Forfeited | | (88,961) | | | $ | 7.71 | |
Unvested RSUs as of March 31, 2026 | | 4,409,497 | | | $ | 13.68 | |
The weighted-average grant date fair value per share of RSUs granted during the three months ended March 31, 2026 and 2025 was $13.38 and $17.66, respectively. The total intrinsic value of RSUs released during the three months ended March 31, 2026 and 2025 was $11.8 million and $14.1 million, respectively. The total fair value of RSUs vested during the three months ended March 31, 2026 and 2025 was $31.1 million and $36.5 million, respectively.
10. Income Taxes
For the three months ended March 31, 2026, the Company recognized income tax expense of $0.1 million, resulting in an effective tax rate of (0.2)%, as compared to income tax expense of $0.2 million, resulting in an effective tax rate of 0.2%, in the same period for 2025. The primary reconciling items between the federal statutory rate of 21.0% for the three months ended March 31, 2026 and the Company’s overall effective tax rate of (0.2)% was the effect of equity compensation and the valuation allowance recorded against the full amount of its net deferred tax assets. The primary reconciling items between the federal statutory rate of 21.0% for the three months ended March 31, 2025 and the Company’s overall effective tax rate of 0.2% was the effect of equity compensation and the valuation allowance recorded against the full amount of its net deferred tax assets.
A valuation allowance is established when it is more likely than not that some portion or all of a deferred tax asset will not be realized. The realization of deferred tax assets depends on the generation of future taxable income during the period in which related temporary differences become deductible. The Company continues to establish a valuation allowance against the full amount of its net deferred tax assets since it is more likely than not that benefits will not be realized, including those benefits created in the current year. This assessment is based on the Company's historical cumulative losses, which provide strong objective evidence that cannot be overcome with projections of income, as well as the fact the Company expects continuing losses in the future.
11. Loss Per Common Share
Basic and diluted loss per common share was calculated as follows:
| | | | | | | | | | | | | | | |
| For the Three Months Ended
March 31, | | |
| (dollars and shares in millions, except per share amounts) | 2026 | | 2025 | | | | |
| Net loss | $ | (57.6) | | | $ | 82.9 | | | | | |
| | | | | | | |
| Weighted average common shares outstanding - basic | 64.0 | | | 72.5 | | | | | |
| Denominator adjustments for diluted EPS: | | | | | | | |
| Number of stock options and RSUs | — | | | 0.2 | | | | | |
| Denominator adjustments for diluted EPS: | — | | | 0.2 | | | | | |
| Diluted weighted average common shares outstanding | 64.0 | | | 72.7 | | | | | |
| | | | | | | |
| Loss per common share | | | | | | | |
| Basic | $ | (0.90) | | | $ | 1.14 | | | | | |
| Diluted | $ | (0.90) | | | $ | 1.14 | | | | | |
Treasury shares are not considered outstanding and are excluded from the calculation of basic and diluted loss per common share.
The Company reported net losses for the three months ended March 31, 2026 and therefore excluded all stock options and RSUs from the calculation of diluted net loss per common share as their inclusion would have had an anti-dilutive effect, as summarized below:
| | | | | | | |
| For the Three Months Ended
March 31, |
| 2026 | | |
| Stock options | 9.4 | | | |
| | | |
| RSUs | 4.4 | | | |
| 13.8 | | | |
12. Commitments and Contingencies
Clinical and Preclinical Development and Licensing Arrangements
From time to time, the Company enters into contracts in the normal course of business with various third parties who support its clinical trials, preclinical research studies and other services related to its development activities. The scope of the services under these agreements can generally be modified at any time, and the agreement can be terminated by either party after a period of notice and receipt of written notice.
In addition, under licensing and related arrangements to which the Company is a party, the Company may be obligated to make milestone payments to third parties. The payment obligations under these arrangements are contingent upon future events, such as achievement of specified milestones or generation of product sales, and the amount, timing and likelihood of such payments are not known.
Yale University License Agreement
In June 2024, the Company entered into an Amended and Restated License Agreement (the “Amended License Agreement”) with Yale pursuant to which the parties amended and restated the license agreement dated July 5, 2013, as amended to date (the "Original Agreement"). In connection with the signing of the Amended License Agreement, the Company made a payment of $14.95 million to Yale in June 2024,
comprising both an upfront payment connected to the Amended License Agreement and an amount related to the collaboration income under the Novartis License Agreement and Novartis Asset Agreement (see Note 3, Research Collaboration and License Agreements, for a description of the agreements) and the Company made another $5.0 million payment to Yale in June 2025 on the first anniversary of signing. Thereafter, the Company will also pay to Yale (1) up to $15.0 million if it secures approval of the first and second royalty products (as defined in the Amended License Agreement), (2) a low single digit percentage royalty on certain, more narrowly defined “collaboration products,” and (3) a lower single digit royalty on its aggregate worldwide net sales of certain newly defined “meaningfully involved products.”
The Company’s obligations under the Original Agreement to pay Yale minimum annual royalties and certain other annual fees have been eliminated and Yale has agreed to release all claims arising previously under the Original Agreement. Other provisions of the Original Agreement remain materially unchanged under the Amended License Agreement, including the requirement to pay to Yale a minimum license maintenance royalty totaling $0.1 million per year until the first sale to a third party of any licensed product, followed by success-based milestones for the first two licensed products for the development of the protein degradation technologies totaling approximately $3.0 million for the first licensed product and approximately $1.5 million for the second licensed product, certain of which milestones have already been satisfied, and low single-digit royalties on aggregate worldwide net sales of certain licensed products, which may be subject to reductions, and subject to minimum royalty payments that range from $0.2 million to $0.5 million.
13. Related Party Transactions
Consulting Agreement
On February 12, 2026, the Company entered into a consulting agreement with John Houston, Ph.D., the Company’s former President and Chief Executive Officer and a current member of its Board of Directors. Under the terms of the agreement, Dr. Houston will provide consulting and advisory services to the Company until March 1, 2027.
Pursuant to the agreement, the Company agreed to (i) pay Dr. Houston a lump sum of $457,000 in March 2026, which amount was equivalent to the amount that Dr. Houston would have received as an employee for a 2025 bonus based on achievement of our 2025 corporate goals as approved by the Company's board of directors, had he continued to be employed by the Company as President and Chief Executive Officer on the date of payment, (ii) reimburse Dr. Houston for up to $27,914 in COBRA health continuation coverage, subject to his election of such coverage, (iii) pay an hourly rate of $500 for services provided in excess of eight hours per month. The vesting of Dr. Houston's previously-granted equity continued pursuant to terms of the relevant grant agreements.
During the three months ended March 31, 2026, the Company recognized expenses totaling $0.5 million related to this agreement, which are reflected in general and administrative expenses in the accompanying unaudited condensed consolidated financial statements.
14. Restructuring Activity
In September 2025, the Company announced an update on its collaboration with Pfizer and further actions to support value creation by optimizing organizational and cost structures and streamlining operations in advance of multiple anticipated upcoming value inflection points, including: further limiting additional expenditures on the vepdegestrant program to support activities required for commercialization readiness and identification, with Pfizer, of a third party for the commercialization and potential further development of vepdegestrant; reducing the Company's workforce by 15% to streamline operations, with the most significant reductions being roles related to vepdegestrant commercialization; and proactively managing pipeline cost by seeking strategic business development opportunities and by identifying further efficiencies across the business. The September 2025 workforce reduction is expected to be completed by the second quarter of 2026.
Components of Restructuring Charges
During the three months ended March 31, 2026, the Company recognized net restructuring related charges of $1.1 million, comprised primarily of non-cash stock compensation, of which $0.3 million of charges are reflected in research and development expenses and $0.8 million are reflected in general and administrative expenses in the accompanying unaudited condensed consolidated financial statements.
The Company's restructuring accrual totaled $1.6 million and $4.4 million as of March 31, 2026 and December 31, 2025, respectively.
15. Segment Information
The Company's operations are organized into one operating and reportable segment focused on the discovery, development and commercialization of therapies that degrade disease-causing proteins. The segment develops protein degradation therapies designed to harness the body's natural protein disposal system to selectively and efficiently degrade and remove disease-causing protein through the Company's PROteolysis TArgeting Chimera (PROTAC) protein degrader platform.
In the second quarter of 2026, the Company announced that the FDA granted approval for VEPPANU™ (vepdegestrant) for the treatment of adults with ER+/HER2-, ESR1-mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endocrine-based therapy. VEPPANU is the first and only FDA-approved PROTAC protein degrader, a type of heterobifunctional protein degrader therapy.
The Company is also progressing multiple product candidates through clinical development programs, including ARV-102, targeting the leucine-rich repeat kinase 2 protein for the treatment of neurodegenerative diseases; ARV-806, targeting Kirsten rat sarcoma G12D protein for cancers with the G12D mutation, including pancreatic, colorectal and non-small cell lung cancers; ARV-393, targeting the B-cell lymphoma 6 protein for the treatment of relapsed/refractory non-Hodgkin Lymphoma; ARV-027, targeting the polyQ-AR in skeletal muscle for the treatment of Spinal-Bulbar Muscular Atrophy, or SBMA, also known as Kennedy's disease. The Company's tangible assets are held in the United States and all of the Company's revenue has been generated in the United States. The Company manages all business activities on a consolidated basis. The Company's chief operating decision maker is the Chief Executive Officer.
The operating segment's revenue is primarily generated through research collaborations and licensing arrangements with pharmaceutical partners. The terms of these agreements contain multiple goods and services which may include (i) licenses, (ii) research and development activities, and (iii) participation in joint research and development steering committees. The terms of these agreements may include non-refundable, upfront license or option fees, payments for research and development activities, payments upon the achievement of certain milestones and royalty payments based on product sales derived from the collaboration. Revenue is recognized ratably over the Company’s expected performance period under each respective arrangement. The Company also generated revenue through the sale of assets based on fair value. The Company does not have intra-entity sales or transfers.
The accounting policies of the operating segment are the same as those described in the Company's Annual Report on Form 10-K for the year ended December 31, 2025 and in Note 2, Summary of Accounting Pronouncements and Significant Accounting Policies. The chief operating decision maker evaluates the performance of the operating segment and allocates resources based on net income/loss that also is reported on the consolidated income statement as net (loss) income. The measure of the operating segment assets is reported on the consolidated balance sheet as total assets.
The chief operating decision maker uses net loss to monitor budget versus actual results and to analyze cash flows in assessing performance of the segment and allocating resources.
The following table summarizes the reportable segment's financial information:
| | | | | | | | | | | | | | | |
| Three Months Ended
March 31, | | |
| (dollars in millions) | 2026 | | 2025 | | | | |
| Revenue | $ | 15.6 | | | $ | 188.8 | | | | | |
| Less: | | | | | | | |
| Research and development expense | | | | | | | |
| | | | | | | |
Vepdegestrant (ARV-471) (*) | 8.9 | | | 24.1 | | | | | |
| ARV-806 | 6.5 | | | 0.9 | | | | | |
| ARV-102 | 5.5 | | | 6.5 | | | | | |
| | | | | | | |
| ARV-393 | 3.7 | | | 2.6 | | | | | |
| Bavdegalutamide (ARV-110) | 0.2 | | | 1.1 | | | | | |
| | | | | | | |
| Other programs | 2.6 | | | 1.7 | | | | | |
| | | | | | | |
| Non program-specific external expense | 9.2 | | | 13.9 | | | | | |
| | | | | | | |
Compensation and related personnel expense
(including stock-based compensation) | 21.3 | | | 36.9 | | | | | |
| Other research and development expense | 2.4 | | | 3.1 | | | | | |
| | | | | | | |
| Total research and development expense | 60.3 | | | 90.8 | | | | | |
General and administrative expense | 19.1 | | | 26.6 | | | | | |
| Other segment expense, net (**) | 0.1 | | | — | | | | | |
| Income tax expense | 0.1 | | | 0.2 | | | | | |
| | | | | | | |
| Plus: | | | | | | | |
| Interest income, net | 6.4 | | | 11.7 | | | | | |
| Segment net (loss) income | $ | (57.6) | | | $ | 82.9 | | | | | |
(*) Includes net reimbursement to and from Pfizer pursuant to the Vepdegestrant (ARV-471) Collaboration Agreement which are accounted for pursuant to ASC 808 and are recorded as an offset or an increase to research and development expenses.
(**) Includes realized gains/ losses on foreign currency transactions and gains/ losses on sale of marketable securities.
During each of the three months ended March 31, 2026 and 2025, the Company recognized depreciation and amortization expense of $0.7 million.
16. Subsequent Events
On May 1, 2026, the Company issued a press release announcing that the FDA has granted approval for VEPPANU™ (vepdegestrant) for the treatment of adults with ER+/ HER2-, ESR1-mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endocrine-based therapy. VEPPANU is the first-and-only FDA-approved PROteolysis TArgeting Chimera (PROTAC) protein degrader, a type of heterobifunctional protein degrader therapy. FDA approval was received in advance of FDA-assigned PDUFA date of June 5, 2026. The Company and its collaborator, Pfizer, remain on track to announce selection of a third party to commercialize VEPPANU.
Pursuant to the Vepdegestrant (ARV-471) Collaboration Agreement, the Company will receive $50.0 million as the Milestone Payment. The Milestone Payment will be offset by certain amounts that the Company will owe to Yale pursuant to the Yale Agreement.
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion and analysis is meant to provide material information relevant to an assessment of the financial condition and results of operations of our company, including an evaluation of the amount and certainty of cash flows from operations and from outside sources, so as to allow investors to better view our company from management’s perspective. You should read the following discussion and analysis of financial condition and results of operations together with our unaudited condensed consolidated financial statements and the related notes appearing elsewhere in this Quarterly Report on Form 10-Q and the consolidated financial statements and the related notes and discussion and analysis of financial condition and results of operations in our Annual Report on Form 10-K for the year ended December 31, 2025 filed on February 24, 2026. This discussion contains forward-looking statements that involve risks and uncertainties. As a result of many factors, such as those set forth in the section titled “Risk Factors” in our Annual Report on Form 10-K for the year ended December 31, 2025, filed on February 24, 2026 and elsewhere in this Quarterly Report on Form 10-Q, our actual results may differ materially from those anticipated in or implied by these forward-looking statements.
Business Overview
Our Business
We are a biotechnology company dedicated to improving the lives of patients suffering from debilitating and life-threatening diseases. Through our PROteolysis TArgeting Chimera, or PROTAC, protein degradation platform, we are pioneering the development of a new class of therapeutics designed to harness the body’s own natural protein disposal system to selectively and efficiently degrade and remove disease-causing proteins. We believe that our targeted protein degradation approach is a novel therapeutic modality that may provide distinct advantages over existing therapies and address a broad range of targets, including historically undruggable proteins, in areas of significant unmet need.
In the past five years, seven of the programs developed using our PROTAC protein degradation platform have progressed to clinical trials in oncology and neurology indications after demonstrating potent and selective protein degradation in our preclinical studies. We believe favorable clinical trial results in our ongoing oncology and neurology programs would further validate our platform as a new therapeutic modality for the potential treatment of diseases caused by dysregulated intracellular proteins.
In the second quarter of 2026, the U.S. Food and Drug Administration, or FDA, approved VEPPANU™ (vepdegestrant) for the treatment of adults with estrogen receptor-positive, or ER+,/human epidermal growth factor receptor 2-negative, or HER2-, estrogen receptor 1, or ESR1, -mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endocrine-based therapy. VEPPANU is the first and only FDA-approved PROTAC protein degrader, a type of heterobifunctional protein degrader therapy. FDA approval was received in advance of the FDA-assigned Prescription Drug User Fee Act, or PDUFA, date of June 5, 2026.
In September 2025, we and Pfizer, Inc. announced our plan to jointly select a third party for the commercialization and potential further development of vepdegestrant. We and Pfizer, remain on track to announce selection of a third party to commercialize VEPPANU.
We are currently also progressing the following product candidates through clinical development programs:
•ARV-102, targeting the leucine-rich repeat kinase 2, or LRRK2, protein for the treatment of neurodegenerative diseases, including progressive supranuclear palsy, or PSP, and Parkinson's disease, or PD;
•ARV-806, targeting Kirsten rat sarcoma, or KRAS, -G12D protein for cancers with the G12D mutation, including pancreatic, colorectal and non-small cell lung cancer;
•ARV-393, targeting the B-cell lymphoma 6, or BCL6, protein for the treatment of relapsed/refractory non-Hodgkin lymphoma, or NHL; and
•ARV-027, targeting the polyglutamine-expanded androgen receptor, or polyQ-AR, in skeletal muscle for the treatment of Spinal-Bulbar Muscular Atrophy, or SBMA, also known as Kennedy's disease.
We are also advancing several preclinical candidates through early stage development, in a broad range of intracellular disease targets, including proteins that currently cannot be addressed by existing small molecule therapies, commonly referred to as “undruggable” or under-drugged targets. These preclinical candidates include ARV-6723 targeting hematopoietic progenitor kinase 1, or HPK1, and a pan-KRAS degrader targeting multiple variants of KRAS while sparing other RAS isoforms.
Our pipeline, which includes an overview of our clinical and preclinical programs, is summarized below.
*The agents in the pipeline graphic above are currently under investigation; their safety and effectiveness for these investigational uses have not been established.
**Upon submission of final chronic toxicology data in non-human primates and FDA clearance to proceed with the Phase 1b clinical trial.
•Defined terms used in pipeline graphic: AR, androgen receptor; BCL6, B-cell lymphoma 6; ER+, estrogen receptor positive; ESR1, estrogen receptor 1, HER2-, human epidermal growth factor receptor 2-negative, HPK1, hematopoietic progenitor kinase 1; I-O, immuno-oncology; KRAS, Kirsten rat sarcoma viral oncogene homolog; LRRK2, leucine-rich repeat kinase 2; mCRPC, metastatic castration resistant prostate cancer; mHSPC, metastatic hormone sensitive prostate cancer; NSCLC, non small cell lung cancer; NDA, new drug application; NHL, non-Hodgkin lymphoma; polyQ, expanded polyglutamine; PSP, progressive supranuclear palsy; SBMA, spinal bulbar muscular atrophy.
•Footnotes included in pipeline graphic: a. Includes relapsed/refractory angioimmunoblastic T-cell lymphoma (AITL) and relapsed/refractory mature B cell NHL; b. Phase 1/2 combination trials with palbociclib, atirmociclib, abemaciclib, ribociclib, samuraciclib, everolimus.
In addition to the programs above and our early-stage collaborations, including with Pfizer and Genentech, Inc. and F. Hoffman-La Roche Ltd., or Genentech, we are conducting exploratory research and development work on multiple other undisclosed targets.
Clinical Stage Programs: ARV-102, ARV-806, ARV-393 and ARV-027
ARV-102: Oral PROTAC LRRK2 Degrader Program
ARV-102 is an investigational, orally bioavailable PROTAC designed to cross the blood-brain barrier and specifically target and degrade LRRK2, which is a large, multi-domain scaffolding kinase with GTPase activity. ARV-102 is our first oral PROTAC protein degrader in clinical development to treat neurodegenerative diseases.
Traditional small molecule inhibitors, or SMIs, only block LRRK2’s kinase activity, and thus only modify disease processes regulated by the LRRK2 kinase. By degrading the entire protein, LRRK2 degraders are designed to eliminate all of the ways LRRK2 interacts with disease pathology: the scaffolding function, GTPase activity, as well as kinase activity. We believe our LRRK2 degraders are particularly well positioned to be
evaluated in neurodegenerative diseases where there are currently no disease modifying therapies available, including:
•PD, where increased LRRK2 expression and activity contributes to neurodegeneration and pathogenesis of PD; and
•PSP, where genetic variations in LRRK2 are associated with PSP progression and accelerated time to death. PSP is a primary tau-driven disease, and tau uptake by human neurons requires LRRK2 activity. Additionally, we have published data associating the tau pathology of PSP with LRRK2-mediated endolysosomal dysfunction.
Preclinical Development
In preclinical studies, ARV-102 was shown to cross the blood-brain barrier and degrade LRRK2 in cerebrospinal fluid, or CSF, in non-human primates, or NHPs. Our preclinical studies also showed that ARV-102 and other similar LRRK2 PROTAC degrader molecules pharmacologically enhanced lysosomal degradative capacity and number, and reduced pathologic forms of tau in vitro and in vivo. We believe the data from our preclinical studies of ARV-102 further support the potential of PROTAC-induced LRRK2 degradation as a treatment for patients with neurodegenerative diseases.
Clinical Development
We have evaluated ARV-102 in Phase 1 clinical trials in healthy volunteers and patients with PD.
•Healthy Volunteers: We initiated the first-in-human Phase 1 clinical trial for ARV-102 in the first quarter of 2024. We completed the single ascending dose, or SAD, and multiple ascending dose, or MAD, cohorts of the ARV-102 Phase 1 clinical trial in healthy volunteers.
•Patients with PD: We completed enrollment in the SAD cohort of the ARV-102 Phase 1 clinical trial in patients with PD in the second quarter of 2025. We received Clinical Trial Application approval in the Netherlands to initiate a multiple dose cohort of the Phase 1 clinical trial in patients with PD in the second quarter of 2025, and we initiated this multiple dose, or MD, cohort in the third quarter of 2025. In the fourth quarter of 2025, we completed enrollment in the multiple dose cohort.
The ARV-102 Phase 1 clinical trial was designed to assess the safety, pharmacokinetics, and pharmacodynamics of orally administered ARV-102 in patients with Parkinson's disease.
In the first quarter of 2026, we presented data from the single-center, randomized, double-blind, placebo-controlled, multiple dose, or MD, cohort of the Phase 1 clinical trial in patients with Parkinson's disease in an oral presentation at the 2026 International Conference on Alzheimer’s and Parkinson’s Diseases and Related Neurological Disorders 2026 in Copenhagen, Denmark. In the MD cohort, patients were randomized to either placebo or multiple oral doses of ARV-102 (20 mg, 40 mg, or 80 mg) for 28 days with follow-up at day 42.
Data presented from the clinical trial included the following:
Safety Profile
•Multiple oral doses of ARV-102 (20 mg, 40 mg, or 80 mg once daily for 28 days) were well tolerated in participants with Parkinson's disease.
•All treatment-emergent adverse events and treatment-related adverse events were mild in severity, with no serious adverse events, discontinuations, or deaths reported.
•No significant changes in lung functions or respiratory symptoms were observed during the 28 days of treatment or during follow-up.
Pharmacokinetic and Pharmacodynamic Evaluation
•ARV-102 levels in CSF increased in a dose-dependent manner after multiple doses, indicating brain penetration.
•The area under the concentration-time curve (AUC0-24) and the maximum plasma concentration (Cmax) after daily dosing increased with dose with a mean terminal plasma half-life (t1/2) of 68 hours.
•ARV-102 achieved peripheral LRRK2 degradation and dose-dependent degradation of LRRK2 in CSF, with approximately 50% or greater degradation observed at all doses by day 14 and maintained through day 28.
•Endolysosomal and neuroinflammatory pathway proteins that are elevated in LRRK2-related Parkinson's disease (e.g., CD68, GPNMB) were reduced with ARV-102.
•Pharmacology and changes in peripheral biomarkers in patients with Parkinson's disease were consistent with observations in healthy volunteers dosed with ARV-102.
Based on the data, we plan to continue investigation of ARV-102 in neurodegenerative diseases associated with LRRK2 and endolysosomal dysfunction. We plan to share additional biomarker data from the Phase 1 clinical trial in patients with PD in the second half of 2026.
We submitted an investigational new drug application, or IND, earlier this year for ARV-102 with the intention of initiating a Phase 1b clinical trial in patients with PSP in the U.S. during first half of 2026. Following the 30-day review period, the FDA requested final data from our chronic toxicology studies in non-human primates prior to authorizing the initiation of the Phase 1b clinical trial in the U.S. in patients with PSP. As a result, the planned Phase 1b clinical trial, in which we have not yet dosed any patients, is on clinical hold and will not begin until we provide these data to the FDA, which we expect will be available in mid-2026. We anticipate the Phase 1b clinical trial in the U.S. to begin in the second half of 2026. We do not expect this to impact our plans for clinical trials in PSP in the EU, and therefore also believe we have the potential to initiate a registrational trial in PSP in late 2026, pending regulatory feedback, which we are planning as a global clinical trial. We continue to evaluate development options for ARV-102 in Parkinson's disease.
ARV-806: Novel PROTAC KRAS G12D Degrader Program
ARV-806 is an investigational novel PROTAC designed to selectively target and degrade mutant KRAS G12D in solid tumors. KRAS is one of the most frequently mutated human oncogenes and G12D is the most common mutation of the KRAS protein. In normal cells, the KRAS protein regulates cell growth and functions as a molecular switch, cycling between a baseline “OFF” state and only turning “ON” when conditions are appropriate for growth. Mutations, including G12D, lock KRAS in the “ON” form, leading to uncontrolled cell growth and cancer. ARV-806 is designed to degrade both the ON and OFF forms of KRAS G12D and by removing this oncogenic protein, has the potential to shut down the constitutive growth signal and lead to death of the cancer cells. We believe ARV-806 has the potential to address high unmet need in solid tumors, such as pancreatic, colorectal and non-small cell lung cancer, or NSCLC, with KRAS G12D mutation.
Preclinical Development
In the preclinical setting, ARV-806 demonstrated high potency and selectivity, with robust antitumor activity through dose-responsive degradation of KRAS G12D in KRAS G12D mutated cancer models, including pancreatic and colorectal models. ARV-806 formed a ternary complex with both the active "ON" and inactive "OFF" forms of KRAS G12D, achieving potent and durable elimination rather than inhibition of the target. As a result, in preclinical studies, ARV-806 achieved in vitro potency more than 25 times greater than clinical stage KRAS G12D "ON" and "OFF" inhibitors and more than 40 times greater than the leading KRAS G12D clinical-stage degrader.
Clinical Development
We filed an IND with the FDA for ARV-806 in the first quarter of 2025 and received a safe-to-proceed letter from the FDA in the second quarter of 2025. We initiated enrollment in a Phase 1 clinical trial of ARV-806 in patients with advanced solid tumors harboring KRAS G12D mutations in the second quarter of 2025 and this trial is currently ongoing.
In the second quarter of 2026, we announced that we had completed dose escalation enrollment of the Phase 1 clinical trial evaluating ARV-806 in patients with solid tumors harboring KRAS G12D mutations. We
plan to initiate enrollment in the dose expansion cohort of the Phase 1 clinical trial of ARV-806 in patients with solid tumors harboring KRAS G12D mutations. We anticipate sharing initial clinical data in patients with solid tumors harboring KRAS G12D mutations in 2026.
ARV-393: Oral PROTAC BCL6 Degrader Program
ARV-393 is an investigational, orally bioavailable PROTAC designed to specifically target and degrade BCL6, a transcriptional repressor and a key regulator of normal B-cell maturation and differentiation processes. Deregulation of BCL6 function (e.g., via chromosomal translocation, mutations) may lead to malignant transformation and development of NHL. Also as a lineage defining transcription factor of T-follicular helper cells, BCL6 has been implicated in nodal T-follicular helper cell lymphoma, or nTFHL, including the angioimmunoblastic type, formerly angioimmunoblastic T-cell lymphoma, or AITL.
We believe that PROTAC-mediated degradation has the potential to address the historically undruggable nature of BCL6 and that ARV-393 PROTAC-mediated degradation of BCL6 may provide an important novel therapeutic option for patients with NHL. Furthermore, we believe current preclinical data suggest that ARV-393 has the potential to be an attractive combination partner for development of novel therapies for lymphoma, including chemo-free combination regimens and/or “all oral” treatment options.
Preclinical Development
We have conducted preclinical studies of ARV-393 alone, in combination with SOC chemotherapy and biologic agents, as well as oral, investigational small molecule inhibitors in high grade and aggressive diffuse large B-cell lymphoma, or DLBCL, and in combination with glofitamab, a CD20xCD3 bispecific antibody and an emerging SOC option for DLBCL, in models of aggressive high grade DLBCL. We believe the totality of our ARV-393 preclinical data provides a compelling rationale to evaluate ARV-393 in combination with bi-specifics, oral pathway inhibitors, and potentially other SOCs in the larger DLBCL indication.
Clinical Development
We initiated the monotherapy cohort of our first-in-human Phase 1 clinical trial of ARV-393 in patients with relapsed or refractory NHL in the second quarter of 2024 and are currently recruiting patients for this clinical trial. This is an open-label, multicenter, Phase 1 dose escalation trial to evaluate the safety, tolerability PK, pharmacodynamics, and preliminary anti-tumor activity of ARV-393 as a single agent in adult patients with relapsed/refractory NHL. We announced in the first quarter of 2026, and reaffirmed in the second quarter of 2026, that there have been multiple responses observed in early cohorts at doses below the predicted effective exposure levels in patients with both B- and T-cell lymphomas in the first-in-human Phase 1 clinical trial. Dose escalation in the trial is ongoing and the safety profile of ARV-393 supports continuing dose escalation. We also believe these early data support an emerging, and differentiated, therapeutic benefit of ARV-393.
We plan to share updated clinical data from the ongoing Phase 1 clinical trial of ARV-393 in patients with relapsed/refractory NHL at a medical congress in the second half of 2026.
In addition, in the second quarter of 2026, we announced the initiation of a combination cohort in the ongoing Phase 1 clinical trial to evaluate ARV-393 in combination with glofitamab as a chemotherapy-free combination approach in patients with DLBCL. Enrollment in this trial is currently ongoing.
ARV-027: Oral PROTAC polyQ-AR Degrader Program
ARV-027 is an investigational, oral, peripherally restricted PROTAC designed to selectively target and eliminate the polyQ-AR in skeletal muscle. ARV-027 is a product candidate specifically selected for potent in vitro reduction of cytosolic and nuclear polyQ-AR and for favorable skeletal muscle exposure following oral administration.
The polyQ-AR protein is the pathogenic driver of spinal bulbar muscular atrophy, or SBMA, a rare, X-linked, genetically defined neuromuscular disease caused by a CAG trinucleotide repeat expansion in the androgen receptor, or AR, gene, causing protein misfolding and leading to progressive degeneration of the neuromuscular system in men. SBMA is also known as Kennedy's disease. SBMA leads to progressive muscle
weakness, dysphagia, and functional decline, and currently has no disease-modifying therapies approved by the FDA or EMA, representing a significant unmet medical need.
In the first quarter of 2026, at the Kennedy's Disease Association conference, we shared preclinical data in an aggressive SBMA mouse model showing that oral ARV-027 degraded polyQ-AR in muscle, led to meaningful functional improvements, and extended survival. We believe ARV-027 has the potential to become the first treatment option for patients with SBMA, where no disease-modifying therapies exist.
We initiated the first-in-human Phase 1 clinical trial in ARV-027 in healthy volunteers in the first quarter of 2026. We plan to continue enrollment in this Phase 1 clinical trial of ARV-027 in healthy volunteers.
Approved Product: VEPPANU™ (vepdegestrant)
VEPPANU™ (vepdegestrant) is an orally bioavailable PROTAC, estrogen receptor degrader approved in the U.S. for use as a monotherapy in the treatment of adults with ER+/HER2-, ESR1-mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endocrine therapy. VEPPANU is the first and only FDA-approved PROTAC protein degrader, a type of heterobifunctional protein degrader therapy.
We have been co-developing vepdegestrant with Pfizer, pursuant to a collaboration agreement that we and Pfizer entered into in July 2021. Pursuant to this agreement, we granted Pfizer worldwide co-exclusive rights to develop and commercialize vepdegestrant, which at that time, was an investigational, oral PROTAC estrogen receptor degrader. We and Pfizer remain on track to announce selection of a third party to commercialize VEPPANU.
Preclinical Development
In preclinical studies, vepdegestrant demonstrated near-complete ER degradation in tumor cells, induced robust tumor shrinkage when dosed as a single agent in multiple ER-driven xenograft models and showed superior anti-tumor activity when compared to a standard of care agent, fulvestrant, both as a single agent and in combination with a cyclin-dependent kinase, or CDK, 4/6 inhibitor.
Clinical Development
We, along with Pfizer, have ongoing clinical trials of vepdegestrant, for which enrollment of patients is complete, which are summarized below.
•TACTIVE-K, a Phase 1b/2 clinical trial of vepdegestrant in combination with Pfizer's cyclin-dependent kinase 4, or CDK4, inhibitor, atirmociclib; and
•TACTIVE-U, a Phase 1b/2 clinical trial of vepdegestrant in combination with multiple targeted therapies including abemaciclib, ribociclib or Carrick Therapeutics, Inc.'s, or Carrick, cyclin-dependent kinase 7, or CDK7, inhibitor, samuraciclib.
We, along with Pfizer, also have several completed clinical trials of vepdegestrant:
•VERITAC-2, a Phase 3 clinical trial of vepdegestrant as a monotherapy, targeting metastatic breast cancer previously treated with endocrine based therapy;
•VERITAC, a Phase 2 dose expansion clinical trial of vepdegestrant as a monotherapy, targeting previously treated metastatic breast cancer;
•TACTIVE-N, a Phase 2 clinical trial of vepdegestrant as a monotherapy in the neoadjuvant setting; and
•TACTIVE-E, a Phase 1 clinical trial of vepdegestrant in combination with everolimus.
Additionally, VERITAC-3 a clinical trial with a study lead-in of vepdegestrant in combination with palbociclib for the treatment of patients with first-line metastatic breast cancer, is ongoing and enrollment of patients is complete. As previously disclosed, VERITAC-3 will not proceed beyond the study lead-in.
VERITAC-2 Clinical Trial, VEPPANU™ (vepdegestrant) FDA Approval
In the first quarter of 2025, we, along with Pfizer, announced positive topline results from the Phase 3 VERITAC-2 clinical trial in the estrogen receptor 1-mutant, or ESR1m, population, and in the second quarter of 2025, we, along with Pfizer announced detailed results from this clinical trial.
In the clinical trial, vepdegestrant, now approved as VEPPANU™, demonstrated a statistically significant and clinically meaningful improvement in progression-free survival, or PFS, among ER+/HER2- advanced and metastatic breast cancer patients with an ESR1 mutation, reducing the risk of disease progression or death by 43% compared to fulvestrant, which is administered via an intramuscular injection. The median PFS, as assessed by blinded independent central review, was 5.0 months with VEPPANU versus 2.1 months with fulvestrant. In the clinical trial, VEPPANU was generally well tolerated in the trial, with a safety profile consistent with what has been observed in previous studies, and mostly low-grade treatment-emergent adverse events, or TEAEs. The three most common TEAEs observed with VEPPANU were fatigue, increased alanine transaminase, and increased aspartate aminotransferase. Detailed results were presented in a late-breaking oral presentation at the American Society of Clinical Oncology, or ASCO, 2025 Annual Meeting and were highlighted in the ASCO press briefing and selected for Best of ASCO, and were also simultaneously published in the New England Journal of Medicine.
Based on the results from VERITAC-2, in the second quarter of 2025, we and Pfizer submitted an NDA to the FDA for vepdegestrant for the treatment of patients with ER+/HER2- ESR1-mutated advanced or metastatic breast cancer previously treated with endocrine-based therapy. This represented the first NDA submitted for a PROTAC. In the third quarter of 2025, we announced that the FDA accepted the NDA for vepdegestrant and assigned a PDUFA action date of June 5, 2026.
In the second quarter of 2026, we announced that the FDA has approved the Company’s new drug application for VEPPANU™ (vepdegestrant) for the treatment of adults with ER+/ HER2-, ESR1-mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endrocrine-based therapy. FDA approval was received in advance of the FDA-assigned PDUFA date of June 5, 2026. We, along with Pfizer, remain on track to announce selection of a third party to commercialize VEPPANU.
In addition, on May 8, 2026, the National Comprehensive Cancer Network® (NCCN®) added vepdegestrant (VEPPANU) to the latest NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Breast Cancer. Vepdegestrant (VEPPANU) was added as a Category 2A treatment option for patients with hormone receptor (HR)-positive/HER2-negative, ESR1-mutated advanced or metastatic breast cancer after at least one line of endocrine therapy + cyclin-dependent kinase (CDK) 4/6 inhibitor.*
*NCCN makes no warranties of any kind whatsoever regarding their content, use, or application and disclaims any responsibility for their application or use in any way.
Preclinical and Other Programs
We have active preclinical programs in neurology and oncology. In 2025 we announced two new product candidate nominees, ARV-027 and ARV-6723. As described above, we initiated a Phase 1 clinical trial for ARV-027 in the first quarter of 2026.
ARV-6723: Oral PROTAC HPK1 Degrader
ARV-6723 is an investigational, preclinical oral PROTAC designed to degrade HPK1 in solid malignancies. Preclinically, ARV-6723 has shown potent, selective HPK1 degradation and strong anti-tumor immune responses with superior tumor control in low- and high- immunogenic murine syngeneic tumor models. In solid tumor malignancies, such as NSCLC, melanoma, and renal cell carcinoma, or RCC, HPK1 acts as a negative regulator in T-cell receptor signaling, contributing to T-cell exhaustion and suppressing antitumor immunity. In addition, HPK1 has a regulatory role in other immune cell types that can be co-opted by tumors, thus enabling these cancers to resist immuno-oncology therapy. Degrading HPK1 and thus eliminating both its kinase and scaffolding functions has the potential to unleash an immune response with potent anti-tumor effects and minimum off-target toxicity.
We presented preclinical data at the Society for Immunotherapy of Cancer annual meeting in the fourth quarter of 2025 that we believe supports the potential of ARV-6723 to provide sustained anti-tumor immune response as a single agent or in combination with standards of care with improved clinical benefits, including
that: ARV-6723, as a single agent, demonstrates anti-tumor efficacy superior to anti-PD1 or a clinical HPK1 inhibitor and combines with anti-PD1 to further enhance response; and ARV-6723 single agent activity outperforms the HPK1 inhibitor and anti-PD-1 efficacy and reinstitutes the tumor microenvironment.
In addition, we presented preclinical data for ARV-6723 at the AACR Immuno-Oncology Conference in the first quarter of 2026 that support clinical investigation of ARV-6723 in patients with solid tumors harboring high- or low-immunogenic tumor microenvironments, or TME, including immune checkpoint inhibitor, or ICI,-resistant tumor settings. This preclinical data showed robust single-agent antitumor and proinflammatory activity in multiple syngeneic tumor models, including those with immunosuppressive TMEs, and showed greater preclinical activity than an investigational HPK1 inhibitor or an anti-PD-1 antibody.
At the AACR Annual Meeting in the second quarter of 2026 we presented preclinical data that demonstrated greater antitumor activity than SOC ICIs or an investigational HPK1 inhibitor. These preclinical data presented showed that ARV-6723, unlike an inhibitor and the ICIs, reverses T-cell exhaustion, reverses the immunosuppressive microenvironment and boosts innate cell immunity in ICI (aPD1 and aCTLA4) resistant models. We believe these preclinical results support future investigation of ARV-6723 alone or in combination with other agents in patients with high- or low-immunogenic tumors.
We plan to initiate a Phase 1 clinical trial of ARV-6723 in patients with advanced solid tumors in mid-2026. Upon initiation of the clinical trial, ARV-6723 will be our first clinical candidate in immuno-oncology.
Pan-KRAS Program
Our preclinical oral pan-KRAS program targets multiple variants of KRAS that drive solid tumors such as PDAC, colorectal cancer, NSCLC, and esophageal cancer, while sparing other RAS isoforms. We believe selectively targeting KRAS for removal may have benefits to tolerability compared with a pan-RAS approach. The poster presented at the 2025 Triple Meeting in the fourth quarter of 2025 showed that orally bioavailable pan-KRAS degraders have been identified that potently degrade multiple variants of KRAS and spare other RAS isoforms. A tool pan-KRAS PROTAC demonstrated robust single-agent activity and superior combination efficacy with immune checkpoint blockade compared with a pan-RAS (ON) inhibitor (seven complete responses compared with two complete responses).
In the first quarter of 2026, at the AACR Special Conference in Cancer Research: RAS Oncogenesis and Therapeutics, we presented preclinical data that demonstrated: robust efficacy in CDX models of pancreatic, colorectal, and lung cancer, greater tumor growth inhibition than a pan-RAS (ON) inhibitor in a KRAS G13D model, and enhanced combination efficacy with immune checkpoint blockade compared with a pan-RAS (ON) inhibitor in a KRAS G12D syngeneic model.
Other Out-licensed or Completed Programs: Luxdegalutamide (ARV-766) and Bavdegalutamide (ARV-110)
We had been developing luxdegalutamide and bavdegalutamide, each an investigational, orally bioavailable, AR degrading PROTAC targeted protein degrader, for the treatment of men with metastatic castration-resistant prostate cancer, or mCRPC. Both luxdegalutamide and bavdegalutamide demonstrated activity in preclinical models of AR overexpression and AR mutations, both common mechanisms of resistance to current standard-of-care agents in men with prostate cancer. We believed that the differentiated PROTAC pharmacology of luxdegalutamide and bavdegalutamide, including their iterative activity, had the potential to translate into significantly improved clinical outcomes over current SOC agents. However, a comparison of clinical data from separate studies of luxdegalutamide and bavdegalutamide showed that luxdegalutamide’s tolerability and efficacy was more promising than that of bavdegalutamide. As a result, early in the fourth quarter of 2023, we determined to prioritize the initiation of a Phase 3 clinical trial with luxdegalutamide in mCRPC instead of the previously planned Phase 3 clinical trial for bavdegalutamide. Clinical trials for bavdegalutamide (ARV-110-101 and ARV-110-103) were completed in the second quarter of 2025.
In the second quarter of 2024, we completed a transaction with Novartis Pharma AG, or Novartis, which comprised a license agreement, or the Novartis License Agreement, and an asset agreement, or the Novartis Asset Agreement. Pursuant to the Novartis License Agreement, we granted Novartis an exclusive worldwide license for the development, manufacture and commercialization of luxdegalutamide, and we completed the transition of our ongoing and planned clinical trials of luxdegalutamide to Novartis in the fourth quarter of 2024.
Pursuant to the Novartis Asset Agreement, we sold Novartis all of our rights, title and interest in our PROTAC protein degrader targeting AR-V7, a splice variant of the AR.
Our Operations
We commenced operations in 2013. Our operations to date have been limited to organizing and staffing our company, business planning, raising capital, conducting discovery and research activities, filing patent applications, identifying potential product candidates, undertaking preclinical studies and clinical trials, establishing arrangements with third parties for collaborations and for the manufacture of initial quantities of our product candidates and preparing for potential commercialization. To date, we have not generated any revenue from product sales and have financed our operations primarily through sales of assets and equity interests, proceeds from our collaborations and a licensing arrangement, grant funding and debt financing. Since inception through March 31, 2026, we raised approximately $1.7 billion in gross proceeds from the sale of assets and equity interests and the exercise of stock options and had received an aggregate of $933.1 million in payments primarily from collaboration partners and a licensing arrangement.
We are a biotechnology company, with product candidates in clinical development and other drug discovery activities in the research and preclinical development stages. Our ability to generate revenue from product sales sufficient to achieve profitability will depend heavily on the successful development and eventual commercialization of one or more of our product candidates.
In the second quarter of 2026, we announced that the FDA has approved VEPPANU™ (vepdegestrant) for the treatment of adults with ER+/HER2-, ESR1-mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endocrine-based therapy. In September 2025, we and Pfizer announced our plan to jointly select a third party for the commercialization and potential further development of vepdegestrant. We, along with Pfizer, remain on track to announce selection of a third party to commercialize VEPPANU. We expect that all decisions related to pricing, access, reimbursement, and ex-U.S. regulatory plans for VEPPANU will be determined by the selected partner.
Any delay or failure to obtain regulatory approvals would materially adversely affect our product candidate development efforts and our business overall. Because of the numerous risks and uncertainties associated with product development, we are unable to predict the timing or amount of increased expenses or when or if we will be able to achieve or maintain profitability. Even if we are able to generate product sales, we may not become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations.
We regularly review our operations and make decisions we believe best support our business strategy. In April 2025, as part of our decision to streamline operations across our organization and enable the efficient progression of our portfolio, we committed to and approved a reduction of our workforce by approximately 33% across all areas of our company. The workforce reduction was aimed at reducing internal costs while minimally impacting our targeted clinical stage programs to drive value over the next several years by aligning our operations with long-term program development objectives. The workforce reduction was substantially completed by the end of the second quarter of 2025.
In September 2025, we announced an update on our collaboration with Pfizer and further actions to support value creation by optimizing organizational and cost structures and streamlining operations in advance of multiple anticipated upcoming value inflection points, including: further limiting additional expenditures on the vepdegestrant program to support activities required for commercialization readiness and identification, with Pfizer, of a third party for the commercialization and potential further development of vepdegestrant; reducing our workforce by an additional 15% to streamline operations, with the most significant reductions being roles related to vepdegestrant commercialization; and proactively managing pipeline cost by seeking strategic business development opportunities and by identifying further efficiencies across the business. The September 2025 workforce reduction is expected to be completed by the second quarter of 2026. Refer to Note 14, Restructuring Activity, in this Quarterly Report on Form 10-Q for further details.
In the first quarter of 2026, we announced the appointment of Randy Teel, Ph.D., as our President, Chief Executive Officer and as a member of our board of directors. Dr. Teel, who previously served as our Chief Business Officer, succeeds John Houston, Ph.D., who is retired from his role as President, Chief Executive Officer, and Chair of Arvinas’ board of directors. Dr. Houston will continue to serve as a member of the Board
and has entered into a consulting agreement with us whereby he will provide consulting and advisory services. Briggs Morrison, M.D., our lead independent director, has been elected to serve as Chair of our board of directors.
Since inception, we have incurred significant operating losses and, even in light of our workforce reductions and cost optimization decisions, expect to continue to incur operating losses for at least the next several years. In addition to any additional costs not currently contemplated due to the events associated with or resulting from our workforce reductions, our ability to achieve profitability and our financial position will depend, in part, on the rate of our future expenditures, potential collaboration revenue, our ability to successfully implement cost avoidance measures and reduce overhead costs and our ability to obtain additional funding.
We expect to continue to incur significant expenses associated with: our ongoing and anticipated preclinical and clinical activities, development activities, research activities in oncology, neuroscience and other disease areas, managing our employees and retaining key talent in research, clinical trials, quality and other functional areas, expenses incurred with contract manufacturing organizations, or CMOs, and contract development and manufacturing organizations, or CDMOs, to supply us with product for our preclinical and clinical studies and expenses incurred with contract research organizations, or CROs, for the synthesis of compounds in our preclinical development activities, as well as other associated costs including those related to partnering with us on our clinical trial portfolio and the management of our intellectual property portfolio.
We do not expect to generate any revenue from product sales in the near future, if ever. While we do have one approved product, VEPPANU, as we announced in September 2025, we and Pfizer have agreed to jointly select a third party for the commercialization and potential further development of VEPPANU. Given this, we do not expect to begin to generate revenue, if any, until after selection of a third party for the commercialization of VEPPANU. Further, we may never generate product revenue from a third party agreement to realize any profits from the out-license of VEPPANU. We are on track to select a third party, and will not know financial terms until the deal is finalized. We expect that all decisions related to pricing, access, reimbursement, and ex-U.S. regulatory plans for VEPPANU will be determined by the selected partner.
Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research or product development programs or any future commercialization efforts, or to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
As of March 31, 2026, we had cash, cash equivalents and marketable securities of $614.9 million. We believe the existing cash, cash equivalents and marketable securities on hand will be sufficient to fund our operations into the second half of 2028, which will enable us to execute on multiple data readouts across our programs. We have based this estimate on assumptions that may prove to be wrong, and we could exhaust our available capital resources sooner than we expect. See “Liquidity and Capital Resources” below.
Financial Operations Overview
Revenue
To date, we have not generated any revenue from product sales and do not expect to generate any revenue from the sale of products in the near future. Our revenues to date have been generated through research collaborations, licensing arrangements and an asset sale. Revenue is recognized ratably over our expected performance period under each agreement.
While we do have one approved product, VEPPANU, as we announced in September 2025, we are planning, with Pfizer, to jointly select a third party for the commercialization and potential further development of VEPPANU. Given this, we do not expect to begin to generate revenue, if any, until after selection of a third party for the commercialization of VEPPANU. Further, we may never generate product revenue from a third party agreement to realize any profits from the out-license of VEPPANU. We and Pfizer remain on track to select a third party, and will not know financial terms until the deal is announced. We expect that all decisions related to pricing, access, reimbursement, and ex-U.S. regulatory plans for VEPPANU will be determined by the selected partner.
We expect that any revenue recognized in the near term will be derived primarily from our current collaboration agreements and licensing arrangement and any additional arrangements that we may enter into in the future. During the year ended December 31, 2025, we received a development milestone totaling $20.0 million, pursuant to the terms of the Novartis License Agreement. To date, no other development, regulatory and commercial milestone payments or royalties have been received under any of our other collaboration agreements or licensing arrangement. However, pursuant to the Vepdegestrant (ARV-471) Collaboration Agreement, the Company will receive $50.0 million as a development milestone payment in connection with the FDA’s approval of VEPPANU, or the Milestone Payment. The Milestone Payment will be offset by certain amounts that the Company will owe to Yale University, or Yale, pursuant to the amended and restated license agreement, dated June 18, 2024, by and between the Company, one of its subsidiaries, and Yale, or the Amended License Agreement.
Pfizer Vepdegestrant (ARV-471) Collaboration Agreement
In July 2021, we entered into the Vepdegestrant (ARV-471) Collaboration Agreement, pursuant to which we granted Pfizer worldwide co-exclusive rights to develop and commercialize products containing our proprietary compound vepdegestrant (ARV-471), or the Licensed Products.
Under the Vepdegestrant (ARV-471) Collaboration Agreement, we received an upfront, non-refundable payment of $650.0 million. In addition, we are eligible to receive up to an additional $1.4 billion in contingent payments based on specified regulatory and sales-based milestones for the Licensed Products. Of the total contingent payments, $400.0 million in regulatory milestones are related to marketing approvals and $1.0 billion are related to sales-based milestones.
We and Pfizer share equally (50/50) all development costs for the Licensed Products (including costs for conducting any clinical trials), subject to certain exceptions.
Unless earlier terminated in accordance with its terms, the Vepdegestrant (ARV-471) Collaboration Agreement will expire on a Licensed Product-by-Licensed Product and country-by-country basis when such Licensed Product is no longer commercialized or developed for commercialization in such country. Pfizer may terminate the Vepdegestrant (ARV-471) Collaboration Agreement for convenience in its entirety or on a region-by-region basis subject to certain notice periods. Either party may terminate the Vepdegestrant (ARV-471) Collaboration Agreement for the other party’s uncured material breach or insolvency. Subject to applicable terms of the Vepdegestrant (ARV-471) Collaboration Agreement, including certain payments to Pfizer upon termination for our uncured material breach, effective upon termination of the Vepdegestrant (ARV-471) Collaboration Agreement, we are entitled to retain specified licenses to be able to continue to exploit the Licensed Products.
Subject to specified exceptions, we and Pfizer have each agreed not to directly or indirectly research, develop, or commercialize any competing products outside of the Vepdegestrant (ARV-471) Collaboration Agreement anywhere in the world during the term of the Vepdegestrant (ARV-471) Collaboration Agreement.
In the second quarter of 2026, we announced that the FDA has granted approval for VEPPANU™ (vepdegestrant) for the treatment of adults with ER+/HER2-, ESR1-mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endrocrine-based therapy. Pursuant to the Vepdegestrant (ARV-471) Collaboration Agreement, we will receive $50.0 million as the Milestone Payment. The Milestone Payment will be offset by certain amounts that the Company will owe to Yale pursuant to the Amended License Agreement.
In September 2025, we announced that we and Pfizer have agreed to jointly select a third party for the commercialization and potential further development of vepdegestrant. The Company and its collaborator, Pfizer, remain on track to announce selection of a third party to commercialize VEPPANU.
Pfizer Research Collaboration Agreement
In December 2017, we entered into a Research Collaboration and License Agreement with Pfizer, setting forth our collaboration to identify or optimize PROTAC targeted protein degraders that mediate for degradation of targets, using our proprietary platform technology that are identified in the agreement or subsequently selected by Pfizer, subject to certain exclusions. We refer to this agreement as the Pfizer Research Collaboration Agreement.
Under the Pfizer Research Collaboration Agreement, Pfizer has designated a number of initial targets. For each identified target protein, we and Pfizer will conduct a separate research program pursuant to a research plan. Pfizer may make substitutions for any of the initial target protein candidates, subject to the stage of research for such target.
In the year ended December 31, 2018, we received an upfront non-refundable payment and certain additional payments totaling $28.0 million in exchange for use of the technology license and to fund Pfizer-related research, as defined within the Pfizer Research Collaboration Agreement. As of March 31, 2026, there remains a single target under the Pfizer Research Collaboration Agreement, and, in accordance with the terms of such Agreement, we are eligible to receive up to an additional $3.8 million in non-refundable option payments if Pfizer exercises such option for the target protein. We are also entitled to receive up to $225.0 million in development milestone payments and up to $550.0 million in sales-based milestone payments for all designated targets under the Pfizer Research Collaboration Agreement, as well as mid- to high-single digit tiered royalties, which may be subject to reductions, on net sales of PROTAC targeted protein degrader-related products.
Novartis Transaction
In April 2024, we entered into a transaction, or the Novartis Transaction, including both a license agreement, or the Novartis License Agreement, and an asset agreement, or the Novartis Asset Agreement, with Novartis Pharma AG, or Novartis. The Novartis Transaction closed in May 2024 upon the expiration of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended, at which time both the Novartis License Agreement and the Novartis Asset Agreement became effective.
Pursuant to the Novartis License Agreement, we granted Novartis an exclusive worldwide license for the development, manufacture and commercialization of luxdegalutamide (ARV-766), our second generation PROTAC AR degrader for patients with prostate cancer. Pursuant to the Novartis Asset Agreement, we sold to Novartis all of our rights, title and interest in our PROTAC protein degrader targeting AR-V7, a splice variant of the AR.
Under the terms of and as consideration for entering into the Novartis Transaction, we received a one-time, upfront payment in the aggregate amount of $150.0 million from Novartis. Under the Novartis License Agreement, we are also eligible to receive up to an additional $1.01 billion as contingent payments based on specified development, regulatory, and commercial milestones for luxdegalutamide (ARV-766) being met, as well as tiered royalties based upon worldwide net sales of luxdegalutamide (ARV-766), subject to reduction under certain circumstances as provided in the Novartis License Agreement. During the year ended December 31, 2025, we received $20.0 million upon the achievement of a development milestone pursuant to the terms of the Novartis License Agreement. There were no development, regulatory or commercial milestone payments, or sales-based royalties received during the three months ended March 31, 2026 and 2025.
The Novartis License Agreement will continue on a country-by-country basis (or, in certain cases, a region-by-region basis) until the expiration of the applicable royalty term for such country (or region, as applicable). The Novartis License Agreement contains customary termination provisions, including that either party may terminate the Novartis License Agreement (a) upon the material breach of the other party or (b) in the event the other party experiences an insolvency event. Additionally, Novartis may terminate the Novartis License Agreement for convenience or upon a safety or regulatory issue.
Genentech License Agreement
In September 2015, we entered into an Option and License Agreement with Genentech focused on PROTAC targeted protein degrader discovery and research for target proteins based on our proprietary platform technology, other than excluded target proteins as described below. This collaboration was expanded in November 2017 through an Amended and Restated Option, License and Collaboration Agreement, which we refer to as the Restated Genentech Agreement. Simultaneous with entering into the Restated Genentech Agreement, Genentech exercised its exclusive option with respect to a PROTAC targeted protein degrader. We receive annual updates on research and development activities related to this option.
Under the Restated Genentech Agreement, Genentech had the right to designate up to ten targets for further discovery and research utilizing our PROTAC platform technology and also had the right to remove a target from the collaboration and substitute a different target that is not an excluded target at any time prior to us
commencing research on such target or in certain circumstances following commencement of research by us. The research phase of the collaboration with Genentech has ended. Genentech is no longer able to nominate new targets into the collaboration. The only Target that remains part of the collaboration is the PROTAC targeted protein degrader for which Genentech exercised its exclusive option for as noted above.
At the time we entered into the original agreement with Genentech, we received an upfront payment of $11.0 million, and at the time we entered into the Restated Genentech Agreement, we received an additional $34.5 million in upfront and expansion target payments. We are eligible to receive payments aggregating up to $44.0 million per target protein upon the achievement of specified development milestones; payments aggregating up to $52.5 million per target protein (assuming approval of two indications) subject to the achievement of specified regulatory milestones; and payments aggregating up to $60.0 million per PROTAC targeted protein degrader directed against the applicable target protein, subject to the achievement of specified sales milestones. These milestone payments are subject to reduction if we do not have a valid patent claim covering the licensed PROTAC targeted protein degrader at the time the milestone is achieved. We are also eligible to receive, on net sales of licensed PROTAC targeted protein degraders, mid-single digit royalties, which may be subject to reductions.
Operating Expenses
Our operating expenses since inception have consisted solely of research and development costs and general and administrative costs.
Research and Development Expenses
Research and development expenses consist primarily of costs incurred for our research activities, including our discovery efforts, and the development of our product candidates, and include:
•employee related expenses, including salaries, benefits, stock-based compensation expense and travel, for personnel engaged in research and development functions;
•expenses incurred under agreements with third parties, including CROs and other third parties that conduct research, preclinical and clinical activities on our behalf as well as third parties that manufacture our product candidates for use in our preclinical studies and clinical trials;
•costs of outside consultants, including their fees, stock-based compensation and related travel expenses;
•the costs of laboratory supplies and developing preclinical studies and clinical trial materials;
•facility-related expenses, which include direct depreciation costs of equipment and allocated expenses for rent and maintenance of facilities and other operating costs;
•costs incurred in the development of intellectual property; and
•third-party licensing fees.
We expense research and development costs as incurred.
We typically use our employee and infrastructure resources across our development programs, and as such, do not track all of our internal research and development expenses on a program-by-program basis. The following table summarizes our research and development expenses for the three months ended March 31, 2026 and 2025:
| | | | | | | | | | | | | | | |
| For the Three Months Ended
March 31, | | |
| (dollars in millions) | 2026 | | 2025 | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Program-specific external expense: | | | | | | | |
Vepdegestrant (ARV-471) (*) | 8.9 | | | 24.1 | | | | | |
| ARV-806 | 6.5 | | | 0.9 | | | | | |
| ARV-102 | 5.5 | | | 6.5 | | | | | |
| | | | | | | |
| ARV-393 | 3.7 | | | 2.6 | | | | | |
| Bavdegalutamide (ARV-110) | 0.2 | | | 1.1 | | | | | |
| | | | | | | |
| Other programs | 2.6 | | | 1.7 | | | | | |
| Total program-specific external expense | 27.4 | | | 36.9 | | | | | |
| Non program-specific external expense | 9.2 | | | 13.9 | | | | | |
| Unallocated internal expense | | | | | | | |
Compensation and related personnel expense
(including stock-based compensation) | 21.3 | | | 36.9 | | | | | |
| Other research and development expense | 2.4 | | | 3.1 | | | | | |
| Total unallocated internal expense | 23.7 | | | 40.0 | | | | | |
| Total research and development expense | $ | 60.3 | | | $ | 90.8 | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
(*) Includes net reimbursement to and from Pfizer pursuant to the Vepdegestrant (ARV-471) Collaboration Agreement which are accounted for pursuant to ASC 808 and are recorded as an offset or an increase to research and development expenses.
Research and development activities are central to our business model. We expect that our research and development expenses will continue to increase substantially for the foreseeable future as we continue to conduct our ongoing clinical trials of ARV-102, ARV-806, ARV-393, ARV-027, and ongoing clinical trials of vepdegestrant, and continue to discover and develop additional product candidates. Research and development expenses related to vepdegestrant have been shared equally with Pfizer since July 22, 2021, the effective date of the Vepdegestrant (ARV-471) Collaboration Agreement. We may receive reimbursement from, or make payments to, Pfizer to satisfy the cost sharing requirements. These payments are accounted for pursuant to ASC 808, Collaborative Arrangements, which are recorded as an offset or an increase to research and development expenses.
We cannot determine with certainty the duration and costs of ongoing and future clinical trials of vepdegestrant, ARV-102, ARV-806, ARV-393, ARV-027 or unexpected costs of ongoing clinical trials for any other product candidate we may develop or if, when, or to what extent we will generate revenue from the commercialization and sale of any product candidate for which we obtain marketing approval.
While we do have one approved product, VEPPANU, as we announced in September 2025, we and Pfizer have agreed to jointly select a third party for the commercialization and potential further development of VEPPANU. We are on track to select a third party, and will not know financial terms until the deal is finalized. We expect that all decisions related to pricing, access, reimbursement, and ex-U.S. regulatory plans for VEPPANU will be determined by the selected partner.
We may never succeed in obtaining marketing approval for any other product candidate.
Further, the successful development and commercialization of our product candidates is highly uncertain. This is due to the numerous risks and uncertainties associated with developing drugs, including the uncertainty of:
•successfully completing preclinical studies and clinical trials;
•receipt and related terms of marketing approvals from applicable regulatory authorities;
•obtaining and maintaining patent and trade secret protection and regulatory exclusivity for our product candidates;
•making or maintaining arrangements with third-party manufacturers, or establishing manufacturing capabilities, for both clinical and commercial supplies of our product candidates;
•establishing sales, marketing, market access and distribution capabilities and launching commercial sales of our products, if and when approved, whether alone or in collaboration with others;
•acceptance of our products, if and when approved, by patients, the medical community and third-party payors;
•obtaining and maintaining third-party coverage and adequate reimbursement;
•maintaining a continued acceptable safety profile of the products following approval; and
•effectively competing with other therapies.
A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA or another regulatory authority were to require us to conduct clinical trials beyond those that we anticipate will be required for the completion of clinical development of a product candidate, or if we experience significant delays in our clinical trials due to patient enrollment or other reasons, we would be required to expend significant additional financial resources and time on the completion of clinical development.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and other related costs, including stock-based compensation for personnel in our executive, finance, business development and administrative functions. General and administrative expenses also include legal fees relating to intellectual property and corporate matters; professional fees for accounting, auditing, tax and consulting services; insurance costs; travel expenses; and facility-related expenses, which include direct depreciation costs and allocated expenses for rent and maintenance of facilities and other operating costs.
We expect that our general and administrative expenses will increase in the future as we manage our personnel, including retaining or hiring of key employees, and, as a result of any future need to increase our headcount to support research and development activities relating to our product candidates, develop our infrastructure and build out commercial operations for any potential launch of commercial sales of our products. We also have incurred and expect to continue to incur expenses associated with being a public company, including costs of accounting, audit, legal, regulatory and tax-related services associated with maintaining compliance with the Nasdaq Stock Market and U.S. Securities and Exchange Commission requirements; director and officer insurance costs; and investor and public relations costs.
Other Income
Other income consists primarily of interest income from marketable securities and money market accounts.
Income Taxes
Since our inception in 2013, we have not recorded any U.S. federal or state income tax benefits for the net losses we have incurred in any year or for our federal or state earned research and development tax credits, due to our uncertainty of realizing a benefit from those items.
As of December 31, 2025, we had $533.6 million of federal net operating loss carryforwards, all of which may be carried forward indefinitely, but the deductibility of such carryforwards is limited to 80% of our taxable income in the year in which carryforwards are used, $563.2 million of state and local net operating loss carryforwards which expire at various dates beginning in 2035, $44.7 million of federal tax credit carryforwards
and $22.3 million of state tax credit carryforwards as of December 31, 2025 which expire at various dates beginning in 2035.
We expect to generate federal and state net operating losses and credit carryforwards in 2026 and future periods. The revenue recognition and capitalization of research expenses are timing differences for tax purposes and deferred tax assets were established. We have provided a valuation allowance against the full amount of the deferred tax assets since, in the opinion of management, based upon our earnings history, it is more likely than not that the benefits will not be realized.
As of March 31, 2026, Arvinas, Inc. had four wholly owned subsidiaries organized as C-corporations: Arvinas Operations, Inc., Arvinas Androgen Receptor, Inc., Arvinas Estrogen Receptor, Inc., and Arvinas Winchester, Inc.
Critical Accounting Policies and Use of Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our unaudited condensed consolidated financial statements, which have been prepared in accordance with generally accepted accounting principles in the United States. The preparation of our unaudited condensed consolidated financial statements and related disclosures requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, costs and expenses and the disclosure of contingent assets and liabilities in our unaudited condensed consolidated financial statements. We base our estimates on historical experience, known trends and events and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions.
There have been no material changes to our critical accounting policies from those described in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our Annual Report on Form 10-K for the year ended December 31, 2025, filed with the Securities and Exchange Commission on February 24, 2026.
Results of Operations
Comparison of the Three Months Ended March 31, 2026 and 2025
| | | | | | | | | | | | | | | | | | | | | | | |
| For the Three Months Ended
March 31, | | | | | | |
| (dollars in millions) | 2026 | | 2025 | | $ change | | | | | | |
| Revenue | $ | 15.6 | | | $ | 188.8 | | | $ | (173.2) | | | | | | | |
| Research and development expenses | (60.3) | | | (90.8) | | | 30.5 | | | | | | | |
| General and administrative expenses | (19.1) | | | (26.6) | | | 7.5 | | | | | | | |
| Other income | 6.3 | | | 11.7 | | | (5.4) | | | | | | | |
| Income tax expense | (0.1) | | | (0.2) | | | 0.1 | | | | | | | |
| | | | | | | | | | | |
| Net (loss) income | $ | (57.6) | | | $ | 82.9 | | | $ | (140.5) | | | | | | | |
Reconciliation of GAAP and Non-GAAP Information | | | | | | | | | | | | | | | | | | | | | |
| For the Three Months Ended
March 31, | | | | |
| (dollars in millions) | 2026 | | 2025 | | | | | | | | |
| Research and development reconciliation | | | | | | | | | | | |
| GAAP research and development expenses | $ | 60.3 | | | $ | 90.8 | | | | | | | | | |
| Less: restructuring expense | 0.3 | | | — | | | | | | | | | |
| Less: stock-based compensation expense (*) | 5.7 | | | 11.5 | | | | | | | | | |
| Non-GAAP research and development expenses | $ | 54.3 | | | $ | 79.3 | | | | | | | | | |
| General and administrative reconciliation | | | | | | | | | | | |
| GAAP general and administrative expenses | $ | 19.1 | | | $ | 26.6 | | | | | | | | | |
| Less: restructuring expense | 0.8 | | | — | | | | | | | | | |
| Less: stock-based compensation (net reversal) expense (*) | 5.3 | | | 3.5 | | | | | | | | | |
| Non-GAAP general and administrative expenses | $ | 13.0 | | | $ | 23.1 | | | | | | | | | |
(*) Excludes restructuring related stock-based compensation. See Note 14, Restructuring Activity, to the unaudited condensed consolidated financial statements for further details.
Non-GAAP Financial Information
We define non-GAAP expenses as GAAP expenses excluding restructuring and stock-based compensation expense. We use the non-GAAP financial measures, non-GAAP research and development expense and non-GAAP general and administrative expense, to evaluate our ongoing operations and for internal planning and forecasting purposes. We believe that non-GAAP financial information, when taken collectively, may be helpful to investors because it provides consistency and comparability with past financial performance. However, non-GAAP financial information is presented for supplemental informational purposes only, has limitations as an analytical tool, and should not be considered in isolation or as a substitute for financial information presented in accordance with GAAP. Other companies, including companies in our industry, may calculate similarly titled non-GAAP measures differently or may use other measures to evaluate their performance, all of which could reduce the usefulness of our non-GAAP financial measures as tools for comparison. Investors are encouraged to review the related GAAP financial measures and the reconciliation of these non-GAAP financial measures to their most directly comparable GAAP financial measures and not rely on any single financial measure to evaluate our business.
Revenue
Revenue for the three months ended March 31, 2026 totaled $15.6 million, compared to $188.8 million for the three months ended March 31, 2025. The decrease of $173.2 million was primarily due to $175.6 million of decreased revenue from the Vepdegestrant (ARV-471) Collaboration Agreement with Pfizer driven by
changes in total program cost estimates recognized in 2025 resulting from the removal of two Phase 3 trials from the development plan, offset by an increase in revenue from the Pfizer Research Collaboration Agreement of $2.3 million due to changes in estimates of the performance period duration recognized in 2025 from updated research timelines.
Research and Development Expenses
Research and development expenses for the three months ended March 31, 2026 totaled $60.3 million, compared to $90.8 million for the three months ended March 31, 2025. The decrease of $30.5 million was primarily due to a decrease in compensation and related personnel expenses of $15.6 million, which are not allocated by program, and a decrease in external expenses of $14.2 million. External expenses include (i) program-specific expenses, which decreased by $9.5 million, primarily driven by decreases in our vepdegestrant (ARV-471) and ARV-102 programs of $15.2 million and $1.0 million, respectively, partially offset by increases in our ARV-806 and ARV-393 programs of $5.6 million and $1.1 million, respectively, and (ii) non-program specific expenses, which decreased by $4.7 million.
Non-GAAP research and development expenses for the three months ended March 31, 2026 totaled $54.3 million, compared to $79.3 million for the three months ended March 31, 2025, excluding $0.3 million of restructuring expense for the three months ended March 31, 2026, and $5.7 million and $11.5 million of non-cash stock-based compensation expense for the three months ended March 31, 2026 and 2025, respectively.
General and Administrative Expenses
General and administrative expenses totaled $19.1 million for the three months ended March 31, 2026, compared to $26.6 million for the three months ended March 31, 2025. The decrease of $7.5 million was primarily due to a decrease in professional fees of $5.3 million and a decrease in costs related to developing our commercial operations of $1.8 million.
Non-GAAP general and administrative expenses for the three months ended March 31, 2026 totaled $13.0 million, compared to $23.1 million for the three months ended March 31, 2025, excluding $0.8 million of restructuring expense for the three months ended March 31, 2026, and $5.3 million and $3.5 million of non-cash stock-based compensation expense for the three months ended March 31, 2026 and 2025, respectively.
Other Income
Other income totaled $6.3 million for the three months ended March 31, 2026, compared to $11.7 million for the three months ended March 31, 2025. The decrease of $5.4 million was primarily due to a decrease in interest income on our marketable securities of $5.3 million.
Income Tax Expense
Income tax expense totaled $0.1 million for the three months ended March 31, 2026, compared to $0.2 million for the three months ended March 31, 2025. The current and prior income tax totals were driven by the effect of equity compensation and the valuation allowance recorded against the full amount of our net deferred tax assets.
Liquidity and Capital Resources
Overview
We have one product, VEPPANU, approved for commercial sale in the United States.
We are planning, with Pfizer, to jointly select a third party for the commercialization and potential further development of VEPPANU. We do not expect to begin to generate revenue, if any, until after selection of a third party for the commercialization of VEPPANU. Further, we may never generate product revenue from a third party agreement to realize any profits from the third party sales of VEPPANU.
To date, we have financed our operations primarily through the sales of assets and equity interests, proceeds from our collaborations and a license arrangement, grant funding and debt financing. Since inception through March 31, 2026, we had received an aggregate of $933.1 million in payments from collaboration partners and a licensing arrangement, grant funding and forgivable and partially forgivable loans from the State
of Connecticut, and raised approximately $1.7 billion in gross proceeds from the sale of assets and equity interests, and the exercise of stock options, including:
•October 2018: completion of our initial public offering in which we issued and sold an aggregate of 7,700,482 shares of common stock, for aggregate gross proceeds of $123.2 million before fees and expenses;
•July 2019: sale of 1,346,313 shares of common stock to Bayer AG for aggregate gross proceeds of $32.5 million;
•November 2019: completion of a follow-on offering in which we issued and sold 5,227,273 shares of common stock for aggregate gross proceeds of $115.0 million before fees and expenses;
•September – December 2020: sale of 2,593,637 shares of common stock in an “at-the-market offering” for aggregate gross proceeds of $65.6 million before fees and expenses;
•December 2020: completion of a follow-on offering in which we issued and sold 6,571,428 shares of common stock for aggregate gross proceeds of $460.0 million before fees and expenses;
•September 2021: issuance of 3,457,815 shares of common stock to Pfizer for aggregate gross proceeds of $350.0 million;
•July - September 2023: sale of 1,449,275 shares of common stock in an “at-the-market offering” for aggregate gross proceeds of $37.2 million before fees and expenses;
•November 2023: sale of 12,963,542 shares of common stock and pre-funded warrants to purchase 3,422,380 shares of common stock in a private placement for aggregate gross proceeds of $350.0 million before fees and expenses; and
•April 2024: sale of AR-V7 to Novartis under the Novartis Asset Agreement for $20.0 million.
In November 2023, we amended and restated the Equity Distribution Agreement with Piper Sandler & Company and Cantor Fitzgerald & Co., pursuant to which we may offer and sell from time to time, through the agents, up to approximately $262.8 million of the common stock registered under our universal shelf registration statement pursuant to one or more “at-the-market" offerings. During the three months ended March 31, 2026, no shares were issued under the amended and restated agreement.
Cash Flows
Our cash, cash equivalents, and marketable securities totaled $614.9 million and $685.4 million as of March 31, 2026 and December 31, 2025, respectively. We had an outstanding loan balance of $0.5 million and $0.6 million as of March 31, 2026 and December 31, 2025, respectively.
The following table summarizes our sources and uses of cash for the period presented:
| | | | | | | | | | | | | | | | | |
| | For the Three Months Ended
March 31, | | |
| (dollars in millions) | 2026 | | 2025 | | $ change |
| Net cash used in operating activities | $ | (69.2) | | | $ | (88.9) | | | $ | 19.7 | |
| Net cash provided by investing activities | 13.7 | | | 69.5 | | | (55.8) | |
| Net cash used in financing activities | (0.1) | | | (0.1) | | | — | |
| Net decrease in cash and cash equivalents | $ | (55.6) | | | $ | (19.5) | | | $ | (36.1) | |
Operating Activities
Net cash used in operating activities for the three months ended March 31, 2026 decreased by $19.7 million, compared with the three months ended March 31, 2025, primarily due to a decrease in deferred revenue of $173.3 million, driven by changes in total Vepdegestrant (ARV-471) Collaboration Agreement program cost in 2025 resulting from the removal of two Phase 3 trials from the development plan, and changes in prepaid expenses and other assets of $2.1 million, partially offset by an increase in our net loss of $140.5 million, a
decrease in non-cash charges of $4.0 million, as well as changes in accounts receivable of $6.1 million, and accounts payable and accrued liabilities of $6.0 million. The change in non-cash charges was primarily due to a decrease in amortization of collaboration contract asset of $3.0 million and a decrease in stock-based compensation of $2.9 million, partially offset by net accretion of bond discounts/premiums of $2.1 million.
Investing Activities
Net cash provided by investing activities for the three months ended March 31, 2026 decreased by $55.8 million, compared with the three months ended March 31, 2025, primarily due to an increase in purchases of marketable securities of $59.3 million and a decrease in maturities of $24.5 million, partially offset by an increase in sales of marketable securities of $28.9 million.
Financing Activities
Net cash used in financing activities for the three months ended March 31, 2026 remained unchanged from the same period in 2025.
Funding Requirements
Since our inception, we have incurred significant operating losses. We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future as we advance the preclinical and clinical development of our product candidates.
Specifically, we anticipate that our expenses will increase substantially if and as we:
•continue our ongoing and planned clinical trials of our product candidates, including ARV-102, our PROTAC protein degrader designed to target the LRRK2 protein, ARV-806, our PROTAC protein degrader designed to target KRAS G12D for mutated cancers, ARV-393, our PROTAC protein degrader designed to target the BCL6 protein, ARV-027, our PROTAC protein degrader designed to target the polyQ-AR protein, and vepdegestrant, for the treatment of patients with locally advanced or metastatic ER+/HER2- breast cancer;
•progress our preclinical programs, including ARV-6723 and our pan-KRAS degrader program;
•progress additional PROTAC protein degrader programs into IND- or CTA-enabling studies;
•continue to work with Pfizer to select a third party to commercialize and develop VEPPANU;
•apply our PROTAC Discovery Engine to advance additional product candidates into preclinical and clinical development;
•expand the capabilities of our PROTAC Discovery Engine;
•seek marketing approvals for any product candidates that successfully complete clinical trials;
•make decisions with respect to our personnel, including retention or future hiring of key employees, and establishment of a sales, marketing, market access, and distribution infrastructure to launch commercial sales of our products, if and when approved, whether alone or in collaboration with others;
•make decisions with respect to our infrastructure and capabilities, including to support our operations as a public company and our research, product development and future commercialization efforts;
•make or maintain arrangements with third-party manufacturers, or establish manufacturing capabilities, for both clinical and commercial supplies of our product candidates; and
•expand, maintain and protect our intellectual property portfolio.
We had cash, cash equivalents and marketable securities totaling approximately $614.9 million as of March 31, 2026. We believe that our cash, cash equivalents and marketable securities as of March 31, 2026 will enable us to fund our planned operating expenses and capital expenditure requirements into the second half of 2028. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital
resources sooner than we currently expect. Our future capital requirements will depend on many factors, including:
•the progress, scope, costs and results of our ongoing and planned clinical trials of ARV-102, ARV-806, ARV-393 and ARV-027, as well as ongoing clinical trials of vepdegestrant;
•the progress, scope, costs and results of preclinical and clinical development for our other product candidates and development programs, including ARV-6723 and our pan-KRAS degrader program;
•the number of, and development requirements for, other product candidates that we pursue, including our other oncology and neurology research programs;
•the success of our collaborations, including with Pfizer and Genentech;
•work to transition the commercialization and further development of VEPPANU to a third party;
•the costs, timing and outcome of regulatory review of our product candidates;
•the costs and timing of future commercialization activities, including product manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive marketing approval and which we choose to commercialize ourselves;
•the revenue, if any, received from commercial sales of our product candidates for which we receive marketing approval;
•the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims; and
•our ability to establish additional collaboration arrangements with other biotechnology or pharmaceutical companies on favorable terms, if at all, or enter into license, marketing and royalty arrangements, and similar transactions for the development or commercialization of our product candidates.
As a result of these anticipated expenditures, we will need to obtain substantial additional financing in connection with our continuing operations. Until such time, if ever, as we can generate substantial revenue from product sales, we expect to finance our cash needs through a combination of equity offerings, debt financings, collaborations, strategic alliances and marketing, distribution or licensing arrangements. Although we may receive potential future payments under our collaborations, including with Pfizer and Genentech and our out-license to Novartis, we do not currently have any committed external source of funds. Adequate additional funds may not be available to us on acceptable terms, or at all. If we are unable to raise capital when needed or on attractive terms, we may be required to delay, limit, reduce or terminate our research, product development programs or any future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, the terms of these securities may include liquidation or other preferences that adversely affect the rights of our common stockholders. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making acquisitions or capital expenditures or declaring dividends.
If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us.
Borrowings
In June 2018, we entered into an additional assistance agreement with the State of Connecticut, or the 2018 Assistance Agreement, to provide funding for the expansion and renovation of laboratory and office space. We borrowed $2.0 million under the 2018 Assistance Agreement in September 2018, of which $1.0 million was forgiven upon meeting certain employment conditions. Borrowings under the agreement bear an interest rate of 3.25% per annum, with interest only payments required for the first 60 months, and mature in September 2028.
The 2018 Assistance Agreement requires that we be located in the State of Connecticut through September 2028 with a default penalty of repayment of the full original funding amount of $2.0 million plus liquidated damages of 7.5% of the total amount of funding received. As of March 31, 2026, $0.5 million remains outstanding under the 2018 Assistance Agreement.
Item 3. Quantitative and Qualitative Disclosures About Market Risk.
We are exposed to market risks in the ordinary course of our business. These risks primarily include interest rate sensitivities. Our interest-earning assets consist of cash, cash equivalents and marketable securities. Interest income earned on these assets totaled $6.4 million and $11.7 million for the three months ended March 31, 2026 and 2025, respectively. Our interest income is sensitive to changes in the general level of interest rates, primarily U.S. interest rates. As of March 31, 2026, our cash equivalents consisted of bank deposits and money market funds, and our marketable securities included interest-earning securities. Such interest earning instruments carry a degree of interest rate risk. Our outstanding debt totaled $0.5 million and $0.7 million as of March 31, 2026 and December 31, 2025, respectively, and carries a fixed interest rate of 3.25% per annum.
Item 4. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer (our principal executive officer and principal financial officer, respectively), evaluated the effectiveness of our disclosure controls and procedures as of March 31, 2026. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by the company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the Securities and Exchange Commission’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and our management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of March 31, 2026, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Changes in Internal Control over Financial Reporting
No change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) occurred during the fiscal quarter ended March 31, 2026 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
PART II—OTHER INFORMATION
Item 1. Legal Proceedings.
From time to time, we may become involved in litigation or other legal proceedings arising in the ordinary course of business and regardless of outcome, litigation can have an adverse impact on our business, financial condition, results of operations and prospects because of defense and settlement costs, diversion of management resources and other factors. We are not currently a party to any material litigation or legal proceedings.
Item 1A. Risk Factors.
Investing in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties discussed in “Part I, Item 1A, Risk Factors,” in our Annual Report on Form 10-K for the year ended December 31, 2025 filed with the U.S. Securities and Exchange Commission, or SEC, on February 24, 2026, together with all of the other information contained in this Quarterly Report on Form 10-Q, including our unaudited condensed consolidated financial statements and the related notes appearing elsewhere in this Quarterly Report on Form 10-Q. New or revised risk factors can emerge from time to time, and it is not possible to predict the impact that any factor or combination of factors may have on our business, prospects, financial condition and results of operations. The risk factor disclosures in our Annual Report on Form 10-K for the year ended December 31, 2025 are qualified by the information that is described in this Quarterly Report on Form 10-Q. If any of the risks in our Annual Report on Form 10-K for the year ended December 31, 2025 actually occur, our business, prospects, operating results and financial condition could suffer materially. In such an event, the trading price of our common stock could decline and you might lose all or part of your investment. The new and revised risks described below and the risks described in our Annual Report on Form 10-K for the year ended December 31, 2025 are not our only risks. Additional risks and uncertainties not currently known to us or that we currently deem to be immaterial also may materially adversely affect our business, financial condition or future results.
New Risk Factor
In addition to the risks included in our Annual Report on Form 10-K for the year ended December 31, 2025, the following risk may also affect our business:
We currently have only one approved product, VEPPANU™ (vepdegestrant), and the success of VEPPANU will depend on our and Pfizer’s ability to identify and successfully execute a commercialization arrangement with a third party and on the terms of any such deal.
In the second quarter of 2026, we announced that the FDA approved our new drug application for VEPPANU™ (vepdegestrant) for the treatment of adults with ER+/ HER2-, ESR1-mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endrocrine-based therapy. VEPPANU is the first and only FDA-approved PROteolysis TArgeting Chimera (PROTAC) protein degrader, a type of heterobifunctional protein degrader therapy.
In the third quarter of 2025, we announced that we and our collaborator, Pfizer, have agreed to jointly select a third party for the commercialization and potential future development of vepdegestrant. In the second quarter of 2026, we announced that, we and Pfizer remain on track to announce selection of a third party to commercialize VEPPANU. We expect that all decisions related to pricing, access, reimbursement, and ex-U.S. regulatory plans for VEPPANU will be determined by the selected partner. We will have no control over these decisions.
While we believe we are on track to announce selection of a third party, there is no assurance that we actually will be able to identify a suitable partner for VEPPANU or that, if we are able to identify such a partner, we will be able to enter into a definitive agreement with that partner on acceptable terms, or at all, or enter into such an agreement in a timely manner. The failure to enter into a definitive agreement for VEPPANU would significantly delay or prevent the further development and commercialization of VEPPANU. Further, a new partner may not adequately fund or perform its obligations to commercialize or further develop VEPPANU or may not achieve desired results in a timely manner. If we and Pfizer are successful in entering into a strategic relationship with a third party, we would be substantially dependent on the resources and expertise of such third party for the commercialization and further development of VEPPANU.
The commercial success of VEPPANU will be dependent upon our ability to enter into an agreement, and close any transaction, with a third party. Failure to enter into an agreement or close a transaction or entering into an agreement where the third party does not perform its obligations, may mean that VEPPANU does not enter the market on a timely basis, or at all.
Amended Risk Factors
The risk listed below, a version of which was included in our Annual Report on Form 10-K for the year ended December 31, 2025, are replaced in their entirety by the following.
We have an ongoing collaboration with Pfizer related to vepdegestrant, but have announced that we and Pfizer have agreed to jointly select a third party for the commercialization and potential future development of vepdegestrant. If our collaboration with Pfizer or another party is not successful, we may not be able to capitalize on the market potential of vepdegestrant.
In July 2021, we entered into a collaboration agreement with Pfizer, or the Vepdegestrant (ARV-471) Collaboration Agreement, pursuant to which we granted Pfizer worldwide co-exclusive rights to develop and commercialize products containing our proprietary compound vepdegestrant, or the Licensed Products. Although pursuant to the terms of the Vepdegestrant (ARV-471) Collaboration Agreement, we and Pfizer share equally (50/50) all development costs, including costs for conducting clinical trials, for the Licensed Products, subject to certain exceptions, our control over the amount and timing of resources that Pfizer dedicates to the development or commercialization of the Licensed Products is limited, including with respect to oversight and management of CMOs, CDMOs and CROs. Our ability to generate revenues from the Vepdegestrant (ARV-471) Collaboration Agreement will depend, in part, on Pfizer’s ability to successfully perform the functions assigned to it in such agreement.
While our agreement with Pfizer is ongoing, in the third quarter of 2025, we announced that we and Pfizer have agreed to jointly select a third party for the commercialization and potential future development of vepdegestrant, now approved as VEPPANU. We cannot predict the success of our collaboration with Pfizer, or any efforts by Pfizer and us to engage a third party for commercialization of vepdegestrant. We cannot guarantee that our collaboration with Pfizer or the engagement of a third party commercialization partner, if we and Pfizer are successful in doing so, will lead to development or commercialization of the Licensed Products in the most efficient manner or at all. In the second quarter of 2026, we announced that the FDA approved our new drug application for VEPPANU for the treatment of adults with ER+/ HER2-, ESR1-mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endocrine-based therapy. In the second quarter of 2026, we announced that, we and our collaborator, Pfizer, remain on track to announce selection of a third party to commercialize VEPPANU.
There is no assurance that we will be able to identify a suitable partner for VEPPANU, or that, if we are able to identify such a partner, we will be able to enter into a definitive agreement with that partner on acceptable terms, or at all, or in a timely manner. The failure to enter into a definitive agreement for VEPPANU would significantly delay or prevent the further development and commercialization of VEPPANU. Further, a new partner may not adequately fund or perform its obligations to commercialize VEPPANU or may not achieve desired results in a timely manner. If we and Pfizer are successful in entering into a strategic relationship with a third party, we would be substantially dependent on the resources and expertise of such third party for the commercialization of VEPPANU.
We have yet to determine how any potential transaction with a third party commercialization partner will impact us under the existing terms of the Vepdegestrant (ARV-471) Collaboration Agreement. Further, we cannot predict the potential terms of any third party arrangement with respect to the commercialization of vepdegestrant. In addition, Pfizer has a right to terminate the Vepdegestrant (ARV-471) Collaboration Agreement for convenience, subject to certain notice periods. As a result of any of the above, we may not receive any of the $1.4 billion in contingent payments based on specified regulatory and sales-based milestones for the Licensed Products under the Vepdegestrant (ARV-471) Collaboration Agreement.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds.
Sales of Unregistered Securities
We did not issue any securities that were not registered under the Securities Act during the three months ended March 31, 2026.
Item 5. Other Information
Director and Officer Trading Arrangements
From time to time, our directors and officers (as defined in Rule 16a-1(f) under the Exchange Act), engage in open-market transactions with respect to our securities, including to satisfy tax withholding obligations when equity awards vest or are exercised, and for diversification or other personal reasons.
Transactions in our securities by directors and officers are required to be made in accordance with our insider trading policy, which requires that the transactions be in accordance with applicable U.S. federal securities laws that prohibit trading while in possession of material nonpublic information. Rule 10b5-1 under the Exchange Act provides an affirmative defense that enables directors and officers to prearrange transactions in our securities in a manner that avoids concerns about initiating transactions while in possession of material nonpublic information.
The following table describes, for the first quarter of 2026, each trading arrangement for the sale or purchase of Company securities adopted or terminated by our directors and officers that is either (1) a contract, instruction or written plan intended to satisfy the affirmative defense conditions of Rule 10b5-1(c) (a “Rule 10b5-1 trading arrangement”) or (2) a “non-Rule 10b5-1 trading arrangement” (as defined in Item 408(c) of Regulation S-K):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Name (Title) | | Action Taken (Date of Action) | | Type of Trading Arrangement | | Nature of Trading Arrangement | | Duration of Trading Arrangement | | Aggregate Number of Securities (1) |
Randy Teel, Ph.D. (President, Chief Executive Officer, and Director) | | 2/27/2026 | | Durable Rule 10b5-1 trading arrangement for sell-to-cover transactions relating to all equity awards that were granted on or after February 26, 2026; and all equity awards granted prior to February 26, 2026 that vest after the relevant cooling off period | | Sale | | Until final settlement of any restricted stock units, or RSUs | | Indeterminable |
Andrew Saik (Chief Financial Officer and Treasurer) | | 2/27/2026 | | Durable Rule 10b5-1 trading arrangement for sell-to-cover transactions relating to all equity awards that were granted on or after February 26, 2026; and all equity awards granted prior to February 26, 2026 that vest after the relevant cooling off period | | Sale | | Until final settlement of any RSUs | | Indeterminable |
Noah Berkowitz, M.D., Ph.D. (Chief Medical Officer) | | 2/27/2026 | | Durable Rule 10b5-1 trading arrangement for sell-to-cover transactions relating to all equity awards that were granted on or after February 26, 2026; and all equity awards granted prior to February 26, 2026 that vest after the relevant cooling off period | | Sale | | Until final settlement of any RSUs | | Indeterminable |
Angela Cacace, Ph.D. (Chief Scientific Officer) | | 2/27/2026 | | Durable Rule 10b5-1 trading arrangement for sell-to-cover transactions relating to all equity awards that were granted on or after February 26, 2026; and all equity awards granted prior to February 26, 2026 that vest after the relevant cooling off period | | Sale | | Until final settlement of any RSUs | | Indeterminable |
David Loomis (VP, Chief Accounting Officer) | | 2/27/2026 | | Durable Rule 10b5-1 trading arrangement for sell-to-cover transactions relating to all equity awards that were granted on or after February 26, 2026; and all equity awards granted prior to February 26, 2026 that vest after the relevant cooling off period | | Sale | | Until final settlement of any RSUs | | Indeterminable |
(1) The number of shares subject to RSUs that will be sold to satisfy applicable tax withholding obligations upon vesting is unknown as the number will vary based on the extent to which vesting conditions are satisfied, the market price of the Company’s common stock at the time of settlement and the potential future grant of additional RSUs subject to this arrangement. This trading arrangement, which applies to RSUs whether vesting is based on the passage of time and/or the achievement of performance goals, provides for the automatic sale of shares that would otherwise be issuable on each settlement date of a RSU in an amount sufficient to satisfy the applicable tax withholding obligation, with the proceeds of the sale delivered to the Company in satisfaction of the applicable tax withholding obligation.
Item 6. Exhibits.
| | | | | | | | | | | |
Exhibit Number | | Description | |
| | | |
| | | |
| 3.1 | | | |
| | | |
| 3.2 | | | |
| | | |
| 10.1+ | | | |
| | | |
| 10.2+ | | | |
| | | |
| 10.3+ | | | |
| | | |
| 31.1* | | | |
| | | |
| 31.2* | | | |
| | | |
| 32.1** | | | |
| | | |
| 32.2** | | | |
| | | |
| 101.INS* | | Inline XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | |
| | | |
| 101.SCH* | | Inline XBRL Taxonomy Extension Schema Document. | |
| | | |
| 101.CAL* | | Inline XBRL Taxonomy Extension Calculation Linkbase Document. | |
| | | |
| 101.DEF* | | Inline XBRL Taxonomy Extension Definition Linkbase Document. | |
| | | |
| 101.LAB* | | Inline XBRL Taxonomy Extension Label Linkbase Document. | |
| | | |
| 101.PRE* | | Inline XBRL Taxonomy Extension Presentation Linkbase Document. | |
| | | |
| 104.00 | | Cover Page Interactive Date File (formatted as Inline XBRL and contained in Exhibit 101). | |
__________________________________________________
* Filed herewith.
** Furnished herewith.
+ Management contract or compensatory plan or arrangement
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| | | | | | | | |
| Arvinas, Inc. |
| | |
Date: May 11, 2026 | By: | /s/ Randy Teel, Ph.D. |
| | Randy Teel, Ph.D. |
| | President and Chief Executive Officer (Principal Executive Officer) |
| | |
Date: May 11, 2026 | By: | /s/ Andrew Saik |
| | Andrew Saik |
| | Chief Financial Officer and Treasurer (Principal Financial Officer) |
| | |
Date: May 11, 2026 | By: | /s/ David K. Loomis |
| | David K. Loomis |
| | Vice President and Chief Accounting Officer
(Principal Accounting Officer) |