CYBIN INC. DOING BUSINESS AS
HELUS PHARMA
ANNUAL INFORMATION FORM
FOR THE YEAR ENDED MARCH 31, 2026
June 29, 2026
GENERAL
In this annual information form (this “AIF”) unless otherwise noted or the context indicates otherwise, references to the “Company”, “we”, “us” and “our” refer to Cybin Inc. doing business as Helus Pharma (“Helus Pharma”) and its subsidiaries.
Effective April 1, 2025, the Company changed its presentation currency from Canadian dollars (“CA$”) to United States dollars (“U.S. dollars” or “USD”) to better reflect the Company’s operations, align with the currency in which the majority of cash-based expenses are denominated, and improve comparability of its financial results with other publicly traded businesses in the industry. The change in presentation currency has been applied in accordance with IFRS Accounting Standards (“IFRS”) as issued by the International Accounting Standards Board (“IASB”), with comparative financial information translated into the new presentation currency.
All financial information in this AIF is prepared in U.S. dollars and using IFRS as issued by the IASB. This AIF is dated June 29, 2026, and applies to the business activities and operations of the Company for the year ended March 31, 2026, unless otherwise indicated. All dollar amounts in this AIF are expressed in thousands of U.S. dollars, except share and per share amounts, or as otherwise indicated. Reference to CA$ is to thousands of Canadian dollars, except share and per share amounts, or as otherwise indicated.
On September 19, 2024, the outstanding common shares in the capital of the Company (the “Common Shares”) were consolidated on the basis of one new Common Share for every 38 existing Common Shares (the “Consolidation”). As a result, all figures related to shares, warrants and options presented in this AIF have been restated retrospectively for all periods to reflect the Consolidation unless otherwise indicated.
On January 5, 2026, the Company started to operate under the registered business name “Helus Pharma”.
See “Corporate Structure – Name, Address and Incorporation”.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This AIF, and certain documents incorporated by reference in this AIF, contain forward-looking information and forward-looking statements within the meaning of Canadian securities legislation (“forward-looking statements”). All statements other than statements of historical fact contained in this AIF and in documents incorporated by reference in this AIF, including, without limitation, those regarding the Company’s future financial position, business strategy, budgets, research and development, plans and objectives of management for future operations, and any statements preceded by, followed by or that include the words “expect,” “likely”, “may,” “will,” “should,” “intend,” or “anticipate,” “potential,” “proposed,” “estimate” and other similar words, including negative and grammatical variations thereof, or statements that certain events or conditions “may” or “will” happen, or by discussions of strategy, are forward-looking statements.
Forward-looking statements and information include, without limitation, the information concerning possible or assumed future results of operations of the Company set out under “General Development of the Business” and “Description of the Business”, including statements regarding:
•assumptions and expectations described in the Company’s critical accounting policies and estimates;
•the Company’s expectations regarding the adoption and impact of certain accounting pronouncements;
•the Company’s expectations regarding the market for proprietary novel serotonergic agonists (“NSAs”);
•the Company’s expectations regarding legislation, regulations and licensing related to the import, export, processing and sale of NSAs;
•the approval of regulatory bodies of NSA substances including HLP003 and HLP004, for the treatment of various health conditions;
•the healthcare industry in Canada, the United States, the Netherlands, the European Union (the “EU”), Ireland and the United Kingdom;
•the ability to enter and participate in international market opportunities;
•the ability to secure inventory through long-term supply contracts or otherwise;
•product diversification and future corporate development;
•anticipated results of research and development;
•production capacity expectations including discussions of plans or potential for expansion of capacity at existing or new facilities;
•expectations with respect to future expenditures and capital activities; and
•statements about expected use of proceeds from fundraising activities.
These statements are not historical facts, but instead represent only the Company’s expectations, estimates and projections regarding future events. These statements are not guarantees of future performance and involve assumptions, risks and uncertainties that are difficult to predict. Therefore, actual results may differ materially from what is expressed, implied or forecasted in such forward-looking statements. Management provides forward-looking statements because it believes they provide useful information to readers when considering their investment objectives and cautions readers that the information may not be appropriate for other purposes. Consequently, all of the forward-looking statements made in this AIF and in documents incorporated by reference in this AIF are qualified by these cautionary statements and other cautionary statements or factors contained herein, and there can be no assurance that the actual results or developments will be realized or, even if substantially realized, that they will have the expected consequences to, or effects on, the Company. These forward-looking statements are made as of the date of this AIF and the Company assumes no obligation to update or revise them to reflect subsequent information, events or circumstances or otherwise, except as required by law.
The forward-looking statements in this AIF and in documents incorporated by reference in this AIF are based on numerous assumptions regarding the Company’s present and future business strategies and the environment in which the Company will operate in the future, including assumptions regarding business and operating strategies, and the Company’s ability to operate on a profitable basis. The Company does not undertake any obligation to update or release any revisions to these forward-looking statements to reflect events or circumstances after the date of this report, except as may be required by law.
Some of the risks which could affect future results and could cause results to differ materially from those expressed in the forward-looking statements contained herein include:
Risks Related to the Company’s Business and Industry:
•limited operating history;
•achieving publicly announced milestones;
•speculative nature of investment risk;
•early stage of the industry and product development;
•regulatory risks and uncertainties;
•risks of operating in Australia and European countries;
•“foreign private issuer” status under U.S. securities laws;
•the Company may lose “foreign private issuer” status in the future;
•plans for growth;
•limited products;
•limited marketing and sales capabilities;
•no assurance of commercial success;
•no profits or significant revenues;
•reliance on third parties for clinical development activities;
•risks related to third party relationships;
•reliance on contract manufacturers;
•safety and efficacy of products;
•clinical testing and commercializing products;
•completion of clinical trials;
•commercial grade product manufacturing;
•nature of regulatory approvals;
•market access and acceptance;
•unfavourable publicity or consumer perception;
•social media;
•biotechnology and pharmaceutical market competition;
•reliance on key executives and scientists;
•employee misconduct;
•business expansion and growth;
•negative results of external clinical trials or studies;
•product liability;
•enforcing contracts;
•product and material recalls;
•distribution and supply chain interruption;
•difficulty to forecast;
•promoting the brand;
•product viability;
•success of quality control systems;
•reliance on key inputs;
•liability arising from fraudulent or illegal activity;
•operating risk and insurance coverage;
•costs of operating as public company;
•management of growth;
•conflicts of interest;
•foreign operations;
•exchange rate fluctuations
•cybersecurity and privacy risk;
•risks related to artificial intelligence;
•environmental regulation and risks;
•legalization of scheduled serotonergic agonists;
•forward-looking statements may prove to be inaccurate;
•effects of inflation;
•political and economic conditions;
•litigation risk;
•application and interpretation of tax laws;
•enforcement of civil liabilities;
•pandemics;
Risks Related to Intellectual Property:
•trademark protection;
•trade secrets;
•patent law reform;
•patent litigation and intellectual property;
•protection of intellectual property;
•third-party licenses;
Financial and Accounting Risks:
•substantial number of authorized but unissued Common Shares (as defined herein);
•dilution;
•negative cash flow from operating activities;
•additional capital requirements;
•lack of significant product revenue;
•estimates or judgments relating to critical accounting policies;
•inadequate internal controls;
Risks related to the Common Shares:
•market for the Common Shares;
•significant sales of Common Shares;
•volatile market price for the Common Shares;
•tax issues; and
•no dividends.
Although the forward-looking statements contained in this AIF are based upon what management currently believes to be reasonable assumptions, the Company cannot assure prospective investors that actual results, performance or achievements will be consistent with these forward-looking statements. In particular, the Company has made assumptions regarding, among other things:
•substantial fluctuation of losses from quarter to quarter and year to year due to numerous external risk factors, and anticipation that we will continue to incur significant losses in the future;
•uncertainty as to the Company’s ability to raise additional funding to support operations;
•the Company’s ability to access additional funding;
•the fluctuation of foreign exchange rates;
•the risks associated with pandemics;
•the risks associated with the development of the Company’s product candidates which are at early stages of development;
•reliance upon industry publications as the Company’s primary sources for third-party industry data and forecasts;
•reliance on third parties to plan, conduct and monitor the Company’s preclinical studies and clinical trials;
•reliance on third party contract manufacturers to deliver quality clinical and preclinical materials;
•the Company’s product candidates may fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or may not otherwise produce positive results;
•risks related to filing investigational new drug applications to commence clinical trials and to continue clinical trials if approved;
•the risks of delays and inability to complete clinical trials due to difficulties enrolling patients;
•competition from other biotechnology and pharmaceutical companies;
•the Company’s reliance on the capabilities and experience of the Company’s key executives and scientists and the resulting loss of any of these individuals;
•the Company’s ability to fully realize the benefits of acquisitions;
•the Company’s ability to adequately protect the Company’s intellectual property and trade secrets;
•the risk of patent-related or other litigation; and
•the risk of unforeseen changes to the laws or regulations in the United States, the United Kingdom, Canada, the Netherlands, Ireland, Poland, Greece, Australia, and other jurisdictions in which the Company operates.
Drug development involves long lead times, is very expensive and involves many variables of uncertainty. Anticipated timelines regarding drug development are based on reasonable assumptions informed by current knowledge and information available to the Company. Every patient treated on future studies can change those assumptions either positively (to indicate a faster timeline to new drug applications and other approvals) or negatively (to indicate a slower timeline to new drug applications and other approvals). This AIF contains certain forward-looking statements regarding anticipated or possible drug development timelines. Such statements are informed by, among other things, regulatory guidelines for developing a drug with safety studies, proof of concept studies, and pivotal studies for new drug application submission and approval, and assumes the success of implementation and results of such studies on timelines indicated as possible by such guidelines, other industry examples, and the Company’s development efforts to date.
In addition to the factors set out above and those identified in this AIF under “Risk Factors”, other factors not currently viewed as material could cause actual results to differ materially from those described in the forward-looking statements. Although Helus Pharma has attempted to identify important risks and factors that could cause actual actions, events or results to differ materially from those described in forward-looking statements, there may be other factors and risks that cause actions, events or results not to be anticipated, estimated or intended. Accordingly, readers should not place any undue reliance on forward-looking statements.
MARKET AND INDUSTRY DATA
This AIF includes market and industry data that has been obtained from third-party sources, including industry publications. The Company believes that the industry data is accurate and that its estimates and assumptions are reasonable, but there is no assurance as to the accuracy or completeness of this data. Third-party sources generally state that the information contained therein has been obtained from sources believed to be reliable, but there is no assurance as to the accuracy or completeness of included information. Although the data is believed to be reliable, the Company has not independently verified any of the data from third-party sources referred to in this AIF or ascertained the underlying economic assumptions relied upon by such sources. The Company does not intend, and undertakes no obligation, to update or revise any such information or data, whether as a result of new information, future events or otherwise, except as, and to the extent required by, applicable Canadian securities laws.
REGULATORY
The Company’s current business focuses on conducting research and development on of next-generation therapeutics including proprietary novel serotonergic agonists, and is focused on developing and commercializing NSAs. No product will be commercialized prior to applicable legal or regulatory approval.
The Canadian and United States federal governments regulate drugs through the CDSA (as defined herein) and the CSA (as defined herein), respectively, which place controlled substances in a schedule. Under the CDSA, certain NSAs that the Company is developing are currently Schedule III drugs under CDSA and Schedule I drugs under the CSA.
In both Canada and the United States, the applicable federal government is responsible for regulating, among other things, the approval, import, sale and marketing of drugs, including any serotonergic agonists, whether natural or novel. The Company does not deal with psychedelic substances except indirectly within laboratory and clinical trial settings conducted within approved regulatory frameworks in order to identify and develop potential treatments for medical conditions and, further, does not have any direct or indirect involvement with illegal selling, production or distribution of any substances in jurisdictions in which it operates.
In the EU, the INCB (as defined herein), a United Nations entity, oversees the enforcement of international restrictions on controlled substances. EU legislation specifically addresses the regulation of precursors or substances used in the illicit production of drugs through Regulation (EC) No. 273/2004 and Council Regulation (EC) No. 111/2005. However, the EU does not classify different narcotic drugs or psychotropic substances directly. Instead, the Council Decision 2005/387/JHA allows for a decision that can mandate EU member states to impose national controls on a drug, aligning with INCB standards.
EU member states have agreed to prohibit the use of DMT, and in limited and specific cases, inter alia for scientific or medical purposes, regulate the use of DMT. There are specific regulatory requirements in each specific and relevant EU member state, similar to regulating the specific regulatory requirements for the approval of clinical trials at an EU member state level. It is noteworthy to mention that the EU is planning to adopt a pharmaceutical legislation package.
The key legislation in the UK includes MDA (as defined herein), and the MDR (as defined herein), and, if a product is a “medicinal product”, by the Human Medicines Regulations 2012. In the UK, certain NSAs, including HLP003, are classified as Class A drugs under the MDA and Schedule 1 drugs under the MDR, meaning they are considered highly dangerous and subject to the strictest controls and penalties. Their legal manufacture, production, possession, and supply require a special licence from the UK Home Office. DMT is similarly classified as a Class A and Schedule 1 drug under these regulations. The manufacturing and marketing of “medicinal products” requires additional authorization and licences from the MHRA (as defined herein).
The Company oversees and monitors compliance with applicable laws in each jurisdiction in which it operates. In addition to the Company’s senior executives and the employees responsible for overseeing compliance, the Company has local counsel engaged in every jurisdiction in which it operates. See “Compliance Program”. Additionally, the Company has received legal opinions or advice in each jurisdiction where it currently operates regarding (a) compliance with applicable regulatory frameworks and (b) potential exposure and implications arising from applicable laws in jurisdictions where the Company has operations or intends to operate.
For these reasons, the Company may be (a) subject to heightened scrutiny by regulators, stock exchanges, clearing agencies and other authorities, (b) susceptible to regulatory changes or other changes in law, and (c) subject to risks related to drug development, among other things. There are a number of risks associated with the business of the Company. See “Risk Factors” herein.
The Company makes no medical, treatment or health benefit claims about the Company’s proposed products. The U.S. Food and Drug Administration (the “FDA”), Health Canada or other similar regulatory authorities have not evaluated claims regarding NSAs. The efficacy of such products have not been confirmed by approved research. There is no assurance that the use of NSAs can diagnose, treat, cure or prevent any disease or condition. Rigorous scientific research and clinical trials are needed. If the Company cannot obtain the approvals or research necessary to commercialize its business, it may have a material adverse effect on the Company’s performance and operations.
GLOSSARY OF TERMS
In addition to terms defined elsewhere in this AIF, the following terms, when used in this AIF, will have the following meanings (unless otherwise indicated):
“2010 Act” has the meaning set out in Description of the Business.
“2023 ATM Program” has the meaning set out in General Development of the Business – Three Year History.
“2023 Base Shelf Prospectus” has the meaning set out in General Development of the Business – Three Year History.
“2023 Distribution Agreement” has the meaning set out in General Development of the Business – Three Year History.
“2025 ATM Program” has the meaning set out in General Development of the Business – Three Year History.
“2025 Distribution Agreement” has the meaning set out in General Development of the Business – Three Year History.
“Adelia” has the meaning set out in Corporate Structure – Name, Address and Incorporation.
“Adelia Shareholders” has the meaning set out in Corporate Structure – Name, Address and Incorporation.
“Adelia Transaction” has the meaning set out in Corporate Structure – Name, Address and Incorporation.
“ADME” means Absorption, Distribution, Metabolism, and Excretion.
“affiliate” means a company that is affiliated with another company as described below. A company is an “affiliate” of another company if:
(a) one of them is the subsidiary of the other, or
(b) each of them is controlled by the same person.
A company is “controlled” by a person if:
(a) voting securities of the company are held, other than by way of security only, by or for the benefit of that person, and
(b) the voting securities, if voted, entitle the person to elect a majority of the directors of the company.
A person beneficially owns securities that are beneficially owned by:
(a) a company controlled by that person, or
(b) an affiliate of that person or an affiliate of any company controlled by that person.
“Amalco” means the company resulting from the amalgamation of Helus Pharma Corp. and Subco pursuant to the Amalgamation.
“Amalgamation” means the amalgamation of Subco and Helus Pharma Corp. pursuant to Section 174 of the OBCA on the terms and subject to the conditions of the Amalgamation Agreement, which resulted in the reverse takeover of the Company.
“Amalgamation Agreement” means the Amalgamation Agreement dated as of June 26, 2020 among Helus Pharma Corp., Clarmin and Subco relating to the Amalgamation, as amended on October 21, 2020, a copy of which is available under the Company’s profile on SEDAR+ at www.sedarplus.ca.
“APPROACH” has the meaning set out in Description of the Business.
“Arrangement” has the meaning set out in Corporate Structure – Name, Address, and Incorporation.
“Arrangement Agreement” has the meaning set out in Corporate Structure – Name, Address, and Incorporation.
“Asset Acquisition” has the meaning set out in General Development of the Business – Three Year History.
“Associate” has the meaning set out in Section 1(1) of the Securities Act (Ontario), RSO 1990, c.S.5.
“August 2023 Offering” has the meaning set out in General Development of the Business – Three Year History.
“August 2023 Units” has the meaning set out in General Development of the Business – Three Year History.
“August 2023 Warrants” has the meaning set out in General Development of the Business – Three Year History.
“BCBCA” means the Business Corporations Act (British Columbia), as amended.
“Board” means the board of directors of Clarmin prior to the Transaction and the board of directors of the Company following the Transaction.
“BTD” has the meaning set out in General Development of the Business – Three Year History.
“Canadian FDA” has the meaning set out in Description of the Business – Stage of Development of Principal Products.
“Cboe Canada” means Cboe Canada Inc.
“CCMO” has the meaning set out in Description of the Business – Regulatory Environment – Europe (Netherlands).
“CDSA” means the Controlled Drugs and Substances Act (Canada).
“cGMP” has the meaning set out in General Development of the Business – Stage of Development of Principal Products.
“Charter” has the meaning set out in Audit Committee.
“CHDR” has the meaning set out in Description of the Business.
“CIPO” has the meaning set out in Risk Factors – Risks Related to Intellectual Property – Patent Law Reform.
“Clarmin” means Clarmin Explorations Inc., as a company existing, prior to the Transaction, under the BCBCA via articles of incorporation dated October 13, 2016, and continued under the OBCA on November 4, 2020 in connection with the Transaction.
“Clarmin Shares” means the authorized common shares in the capital of Clarmin.
“Class B Share” has the meaning set out in General Development of the Business – Intercorporate Relationships.
“Clinilabs” has the meaning set out in General Development of the Business – Three Year History.
“Closing” has the meaning set out in General Development of the Business – Three Year History.
“CMC” has the meaning set out in Description of the Business – Stage of Development of Principal Products.
“CMDh” has the meaning set out in Description of the Business – Regulatory Environment – Europe (Netherlands).
“CMOs” has the meaning set out in Risk Factors - Reliance on Contract Manufacturers.
“CNS” has the meaning set out in Description of the Business.
“Code” has the meaning set out in Insider Trading Policy and Code of Ethics And Business Conduct – Code of Business Conduct.
“Common Shares” has the meaning set out in General.
“Company” means Cybin Inc., doing business as Helus Pharma, a company existing under the OBCA.
“Consolidation” has the meaning set out in Corporate Structure – Name, Address and Incorporation.
“Contribution Agreement” has the meaning set out in General Development of the Business – Three Year History.
“Court” has the meaning set out in General Development of the Business – Significant Acquisitions and Dispositions.
“CSA” means the Controlled Substances Act (21 U.S.C. § 811, et. seq.).
“CSE” means the Canadian Securities Exchange.
“CTA” means a Clinical Trial Application.
“CTAG” has the meaning set out in Description of the Business – Regulatory Environment – EU (Netherlands).
“CTR” has the meaning set out in Description of the Business – Regulatory Environment – EU (Netherlands).
“Cybin Ireland” means Cybin IRL Limited, a corporation existing under the laws of Ireland and a wholly-owned subsidiary of Helus Pharma Corp..
“DEA” has the meaning set out in General Development of the Business – Three Year History.
“DMT” means N, N-dimethyltryptamine.
“dDMT” has the meaning set out in General Development of the Business – Three Year History.
“Dutch Opium Act” has the meaning set out in Description of the Business – Regulatory Environment – Europe (Netherlands).
“Equity Incentive Plan” means the Company’s omnibus equity incentive plan adopted by the Board on November 5, 2020.
“EMA” has the meaning set out in Description of the Business – Regulatory Environment – Europe (Netherlands).
“EMBRACE” has the meaning set out in Description of the Business.
“Entheon” has the meaning set out in General Development of the Business – Three Year History.
“EU” has the meaning set out in Cautionary Note Regarding Forward-Looking Information.
“Exchange Act” has the meaning set out in General Development of the Business – Three Year History.
“EXTEND” has the meaning set out in Description of the Business.
“FCA” has the meaning set out in Description of the Business – Regulatory Environment – United States.
“FDA” has the meaning set out in Regulatory.
“FFDCA” has the meaning set out in Description of the Business – Stage of Development of Principal Products.
“forward-looking statements” has the meaning set out in Cautionary Note Regarding Forward-Looking Information.
“GAD” has the meaning set out in General Development of the Business – Three Year History.
“GDP” has the meaning set out in Description of the Business – Regulatory Environment - United Kingdom.
“GLP” has the meaning set out in General Development of the Business – Stage of Development of Principal Products.
“GMP” has the meaning set out in Description of the Business – Stage of Development of Principal Products.
“Helus Pharma Corp.” means Helus Pharma Corp., formerly Cybin Corp., prior to giving effect to the Transaction, a corporation existing under the OBCA, which, pursuant to the Transaction, amalgamated with Subco to form Amalco under the name “Cybin Corp.” and became a wholly-owned subsidiary of the Company.
“Helus US” means Helus US Inc., formerly Cybin U.S. Holdings Inc.
“HPFB” has the meaning set out in Description of the Business – Regulatory Environment – Canada.
“IFRS” means International Financial Reporting Standards, as adopted by the International Accounting Standards Board, as amended from time to time.
“IMPD” has the meaning set out in Description of the Business – Regulatory Environment – EU (Netherlands).
“IMP” has the meaning set out in Description of the Business – Regulatory Environment – United Kingdom.
“IM” has the meaning set out in Description of the Business.
“INCB” has the meaning set out in Description of the Business – Regulatory Environment – EU (Netherlands)
“IND” has the meaning set out in General Development of the Business – Three Year History.
“including” means including without limitation, and “include” and “includes” each have a corresponding meaning.
“Insiders” has the meaning set out in Insider Trading Policy and Code of Ethics and Business Conduct.
“Interim Order” has the meaning set out in General Development of the Business – Significant Acquisitions and Dispositions.
“IP” has the meaning set out in Description of the Business.
“IRB” has the meaning set out in General Development of the Business – Three Year History.
“Ireland MDA” has the meaning set out in Research and Development – Ireland.
“Ireland MDR” has the meaning set out in Research and Development – Ireland.
“IV” has the meaning set out in General Development of the Business – Three Year History.
“Listing Statement” means the Cboe Canada Form 1 Listing Statement dated November 9, 2020, as filed on SEDAR+ November 9, 2020, which has been filed as required in accordance with the policies of Cboe Canada.
“LottoGopher” has the meaning set out in Corporate Cease Trade Orders or Bankruptcies; Penalties or Sanctions; Personal Bankruptcies.
“LPC” has the meaning set out in General Development of the Business – Three Year History.
“LPC Purchase Agreement” has the meaning set out in General Development of the Business – Three Year History.
“MADRS” means the Montgomery-Asberg Depression Rating Scale.
“March 2024 Agency Agreement” has the meaning set out in General Development of the Business – Three Year History.
“March 2024 Offering” has the meaning set out in General Development of the Business – Three Year History.
“May 2023 Prospectus” has the meaning set out in General Development of the Business – Three Year History.
“MDA” has the meaning set out in Description of the Business – Regulatory Environment – United Kingdom.
“MDD” has the meaning set out in General Development of the Business – Three Year History.
“MDR” has the meaning set out in Description of the Business – Regulatory Environment – United Kingdom.
“MHRA” has the meaning set out in Description of the Business – Regulatory Environment – United Kingdom.
“MIA(IMP)” has the meaning set out in Description of the Business – Regulatory Environment – United Kingdom.
“Mindset” has the meaning set out in General Development of the Business – Three Year History.
“Natures Journey” means Natures Journey Inc., an Ontario corporation incorporated as a wholly-owned subsidiary of the Company.
“NDA” has the meaning set out in Description of the Business – Non-Revenue Generating Projects.
“NDS” has the meaning set out in Research and Development – Canada.
“NI 51-102” means National Instrument 51-102 Continuous Disclosure Obligations of the Canadian Securities Administrators.
“NI 52-109” means National Instrument 52-109 – Certification of Disclosure in Issuers’ Annual and Interim Filings.
“NI 52-110” means National Instrument 52-110 – Audit Committees.
“November 2023 Offering” has the meaning set out in General Development of the Business – Three Year History.
“November 2023 Units” has the meaning set out in General Development of the Business – Three Year History.
“November 2023 Warrants” has the meaning set out in General Development of the Business – Three Year History.
“NSAs” has the meaning set out in Cautionary Note Regarding Forward-Looking Information.
“NYSE American” has the meaning set out in General Development of the Business – Three Year History.
“OBCA” means the Business Corporations Act (Ontario), as amended.
“Option” means an option to purchase Common Shares granted pursuant to the Equity Incentive Plan.
“Order” has the meaning set out in Corporate Cease Trade Orders or Bankruptcies; Penalties or Sanctions; Personal Bankruptcies.
“OTCQB” has the meaning set out in General Development of the Business – Three Year History.
“PCT” has the meaning set out in General Development of the Business – Three Year History.
“PD” means pharmacodynamic.
“PDD” has the meaning set out in Risk Factors - Risks Related To The Company’s Business and Industry - Early Stage of the Industry and Product Development.
“PIPEDA” has the meaning set out in Risk Factors - Cybersecurity and Privacy Risk.
“Reverse Takeover” has the meaning set out in NI 51-102.
“Rights Plan” has the meaning set out in Description of Capital Structure.
“RMS” has the meaning set out in Description of the Business – Research and Development – EU (Netherlands).
“SAP” has the meaning set out in Description of the Business – Regulatory Environment – Canada.
“SEC” has the meaning set out in General Development of the Business – Three Year History.
“Section 56 Exemption” has the meaning set out in Description of the Business – Regulatory Environment – Canada.
“Serenity Life” means Serenity Life Sciences Inc., an Ontario corporation incorporated as a wholly-owned subsidiary of the Company.
“Small Pharma” has the meaning set out in Corporate Structure – Name, Address and Incorporation.
“Small Pharma Share” has the meaning set out in General Development of the Business – Significant Acquisitions and Dispositions.
“SSRIs” means selective serotonin reuptake inhibitors.
“Subco” means 2762898 Ontario Inc., a wholly-owned subsidiary of Clarmin, incorporated for the purposes of effecting the Amalgamation.
“Support Agreement” has the meaning set out in Corporate Governance – Intercorporate Relationships.
“TPD” has the meaning set out in Description of the Business – Regulatory Environment – Canada.
“Transaction” means the three-cornered amalgamation among Clarmin, Helus Pharma Corp. and Subco pursuant to the terms of the Amalgamation Agreement, which constituted a Reverse Takeover of Clarmin by Helus Pharma Corp.
“TSXV” means the TSX Venture Exchange.
“UN” means the United Nations.
“United Kingdom” or “UK” means the United Kingdom of Great Britain and Northern Ireland.
“United States” or “U.S.” means the United States of America, its territories and possessions, any state of the United States and the District of Columbia.
“USPTO” means the U.S. Patent and Trademark Office.
“Warrant Indenture” has the meaning set out in General Development of the Business – Three Year History.
“Warrants” means warrants to purchase Common Shares.
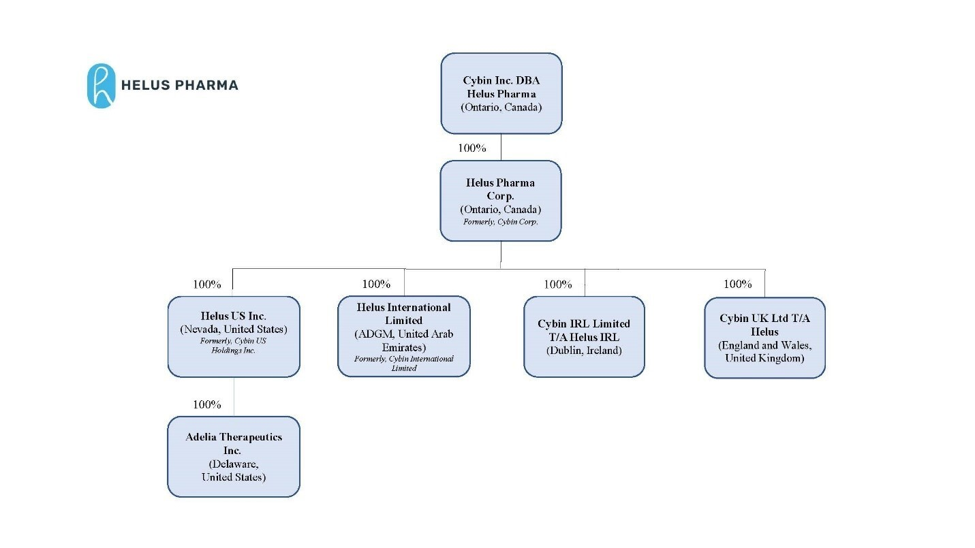
CORPORATE STRUCTURE
Name, Address and Incorporation
The Company (then Clarmin) was incorporated under the BCBCA on October 13, 2016 under the name “Clarmin Explorations Inc.”. On January 8, 2018, the Company completed its initial public offering of Common Shares, pursuant to which the Company issued 3,500,000 Common Shares at a price of CA$0.10 per Common Share (pre-Consolidation) for gross proceeds of CA$350. The Common Shares were listed on the TSXV on January 8, 2018 under the symbol “CX”.
Subco was incorporated under the OBCA on June 26, 2020 for the purposes of effecting the Amalgamation.
On November 2, 2020, in connection with the Transaction, Clarmin consolidated its outstanding Clarmin Shares on a 6.672 old for one (1) new basis.
Upon closing of the Transaction, on November 5, 2020: (i) the Company (then Clarmin) and Helus Pharma Corp. completed a series of transactions resulting in a reorganization of Helus Pharma Corp. and the Company and pursuant to which the Company became the direct parent and sole shareholder of Helus Pharma Corp.; (ii) the Company changed its year end from July 31 to March 31; and (iii) the Company was continued under the OBCA by Certificate and Articles of Continuance and changed its name to “Cybin Inc.”
The Transaction constituted a Reverse Takeover of the Company by Helus Pharma Corp., with Helus Pharma Corp. as the reverse takeover acquirer and the Company as the reverse takeover acquiree, under applicable securities laws and for accounting purposes under IFRS.
The Clarmin Shares were listed on the TSXV until November 5, 2020 when they were delisted from the TSXV in connection with the completion of the Transaction. The Common Shares commenced trading on Cboe Canada on November 10, 2020, under the symbol “CYBN”.
On March 8, 2021, the Company announced that its Common Shares had commenced trading on the OTCQB® Venture Market (the “OTCQB”) under the symbol “CLXPF”.
On August 5, 2021, the Common Shares commenced trading on the NYSE American LLC stock exchange (the “NYSE American”) under the symbol “CYBN”. Concurrent with the commencement of trading on the NYSE American, the Common Shares ceased to be quoted on the OTCQB.
On October 23, 2023, the Company announced the completion of the acquisition by Helus Pharma Corp. of Small Pharma Inc. (“Small Pharma”) by way of a statutory plan of arrangement under the provisions of the BCBCA (the “Arrangement”). The Arrangement was completed pursuant to the terms of an arrangement agreement entered into between the Company and Small Pharma dated August 28, 2023 (the “Arrangement Agreement”). As a result of the Arrangement, Small Pharma became a wholly-owned subsidiary of Helus Pharma Corp.. For further information see “General Development of the Business – Significant Acquisitions and Dispositions”.
On September 19, 2024, the Company completed the Consolidation. As a result, all figures related to shares, warrants and options presented in this AIF have been restated retrospectively for all periods to reflect the Consolidation unless otherwise indicated.
On January 5, 2026, the Company transferred its U.S. stock exchange listing from NYSE American (previous ticker: CYBN) to Nasdaq Global Market (“Nasdaq”) under the ticker symbol “HELP”. The Company continues to be listed on Cboe Canada under the same “HELP” ticker symbol. Concurrent with the commencement of trading on Nasdaq, the Company announced it will be doing business as Helus (pronounced “Heal Us”) Pharma and started to operate under the registered business name “Helus Pharma”. The Company expects to seek approval from shareholders to change its legal name to Helus Pharma Inc. at the Company’s next annual and special meeting of shareholders. Furthermore, the following subsidiaries have changed their legal names:
| | | | | | | | |
Prior Name | New Name | Effective Date |
| Cybin US Holdings Inc. | Helus US Inc. | January 2, 2026 |
| Cybin Corp. | Helus Pharma Corp. | January 5, 2026 |
| Cybin International Limited | Helus International Limited | January 6, 2026 |
The Company’s registered office and head office is located at 100 King Street West, Suite 5600, Toronto, Ontario, M5X 1C9.
Intercorporate Relationships
Helus Pharma Corp. was incorporated under the OBCA on October 22, 2019. Pursuant to the Amalgamation, Helus Pharma Corp. amalgamated with Subco to form Amalco under the name “Cybin Corp.”, which is a wholly-owned subsidiary of the Company. On January 5, 2026, “Cybin Corp.” changed its legal name to “Helus Pharma Corp.” See “Corporate Structure – Name, Address and Incorporation”.
Natures Journey, a wholly-owned, subsidiary of the Company, was formed under the OBCA on November 6, 2019. Effective June 4, 2025, the Company completed the voluntary dissolution of Natures Journey under the OBCA. This entity was non-operational prior to its dissolution.
Serenity Life, a wholly-owned, subsidiary of the Company, was formed under the OBCA on November 6, 2019. Effective June 4, 2025, the Company completed the voluntary dissolution of Serenity Life under the OBCA. This entity was non-operational prior to its dissolution.
Helus US, a fully-controlled subsidiary of the Company, was formed under the law of the State of Nevada on December 4, 2020. Certain of the Company’s business operations pertaining to NSA research and development are conducted through Helus US. On January 2, 2026, Helus US changed its legal name to “Helus US Inc.”
On December 4, 2020, the Company entered into a contribution agreement, as amended on September 24, 2021 (the “Contribution Agreement”) with Helus Pharma Corp., Helus US (formerly Cybin US Holdings Inc.) and all of the shareholders (the “Adelia Shareholders”) of Adelia Therapeutics Inc. (“Adelia”) whereby Helus US agreed to purchase from the Adelia Shareholders all of the issued and outstanding Adelia shares in exchange for the Class B Shares (as defined herein) (the “Adelia Transaction”). Under the Contribution Agreement, the Adelia Shareholders are entitled to Class B Shares upon the occurrence of certain milestones, as set out in the Contribution Agreement. Pursuant to the Contribution Agreement and the support agreement entered into among Helus US and the Adelia Shareholders (the “Support Agreement”), the Adelia Shareholders received 868,833 non-voting Class B common shares in the capital of Helus US (each a “Class B Share”), which are exchangeable for
Common Shares, on a 0.26316 Common Shares for 1 Class B Share basis, at the option of the holder thereof, subject to customary adjustments.
Cybin Ireland, a wholly-owned subsidiary of Helus Pharma Corp., was formed under the Companies Act of 2014 in the country of Ireland on May 6, 2021. In connection with the formation of Cybin Ireland, the Company transferred its intellectual property assets to this entity. In addition, certain of the Company’s business operations, including European operations and research activities with various academic and clinical research organizations, are conducted through Cybin Ireland.
On October 23, 2023, the Company announced the completion of the acquisition of Small Pharma by way of the Arrangement. As a result of the Arrangement, Small Pharma is now a wholly-owned subsidiary of Helus Pharma Corp. On April 1, 2024, pursuant to the provisions of the OBCA, Small Pharma completed a horizontal amalgamation with Helus Pharma Corp., with Helus Pharma Corp. being the resulting entity. As a result of this amalgamation, Cybin UK Ltd. T/A Helus is now a wholly-owned subsidiary of Helus Pharma Corp.
On September 1, 2025, the Company incorporated Helus International Limited (formerly Cybin International Limited), a wholly owned subsidiary of Helus Pharma Corp.
The following chart sets out all the Company’s subsidiaries as at the date hereof, their jurisdictions of incorporation and the Company’s direct and indirect voting interest in each of these subsidiaries.
GENERAL DEVELOPMENT OF THE BUSINESS
On November 5, 2020, Helus Pharma Corp. completed its Reverse Takeover of the Company (then Clarmin) pursuant to the terms of the Amalgamation Agreement. The Transaction was completed by way
of a “three-cornered” amalgamation pursuant to the provisions of the OBCA whereby Helus Pharma Corp. amalgamated with Subco to form an amalgamated corporation and a wholly owned subsidiary of the Company. With the completion of the Transaction the Common Shares became listed for trading on Cboe Canada under the trading symbol “CYBN” and were delisted from the facilities of the TSXV. On January 5, 2026, the Company began trading under the trading symbol “HELP”. See “Corporate Structure – Name, Address and Incorporation”.
The Company is a clinical-stage pharmaceutical company committed to helping minds heal by developing proprietary NSAs. Serotonergic agonists broadly refer to compounds that activate serotonin receptors and include a wide range of approved and investigational drugs with varying selectivity and mechanisms of action. NSAs are a proprietary subset of serotonergic agonists that are synthetically engineered to selectively activate specific serotonin receptor subtypes and signaling pathways, with the aim of achieving differentiated and more precise therapeutic effects.
Helus Pharma’s research and development work focuses on a three-pillar strategy that leverages the Company’s core competencies in preclinical innovation and clinical development. This strategy supports the creation of intellectual property (“IP”) focused on developing the Company’s platform technology to develop NSAs, the progression of clinical development programs studying certain of these NSAs, including HLP003 (previously referred to as CYB003), a deuterated psilocin molecule (“HLP003”), HLP004 (previously referred to as CYB004), a deuterated version of DMT (“HLP004”), HLP005 (previously referred to as CYB005), phenethylamine and tryptamine derivatives (together “HLP005”), and an expansive list of preclinical molecules to facilitate future drug development opportunities. These name changes did not alter the underlying scientific foundations, development plans, or regulatory status of the programs, but were implemented to provide greater consistency, clarity, and alignment with the Helus Pharma brand as the Company advances its pipeline through clinical development and prepares for potential commercialization.
Additional details regarding the Transaction and the business of the Company can be found in the Listing Statement as filed on SEDAR+ on November 9, 2020.
Three Year History1
Year ended March 31, 2024
On April 12, 2023, the Company announced the launch of EMBARK Open Access, an online foundational training course that offers psychedelic facilitation training for healthcare professionals and people interested in offering psychological support.
On May 9, 2023, the Company announced the completion of dosing the last subject in Part B of the Phase 1 HLP004-E trial. With the completion of Part B, the Company announced on May 24, 2023 that it initiated dosing of HLP004 in Part C which will evaluate IV bolus + infusion regimens of HLP004, in a crossover design. Results from Parts B and C are expected to provide a more robust pharmacokinetics and PD model to optimize dose selection and formulation development for future clinical studies. The Company expects to report top-line results from the completed Phase 1 HLP004-E clinical trial in the third quarter of calendar year 2023.2
1 All quarter references in this section are based on calendar year-end.
2 See “Risk Factors” for further information.
On May 30, 2023, the Company announced that it has entered into a common share purchase agreement (the “LPC Purchase Agreement”) with Lincoln Park Capital Fund, LLC (“LPC”). Subject to the terms and conditions of the LPC Purchase Agreement, the Company has the right to sell, and LPC is obligated to purchase, up to $30 million of Common Shares over a 36-month period at prices that are based on the market price at the time of each sale to LPC. The Company, in its sole discretion, controls the timing and amount of all sales of Common Shares under the LPC Purchase Agreement. The sale of Common Shares under the LPC Purchase Agreement will be made pursuant to and qualified by way of a prospectus supplement dated May 30, 2023 (the “May 2023 Prospectus”), to the Company’s short form base shelf prospectus dated July 5, 2021 filed with the securities commissions in each of the provinces and territories of Canada. The May 2023 Prospectus was also filed with the Securities and Exchange Commission (“SEC”) as part of a registration statement on Form F-10, which was declared effective by the SEC on October 8, 2021, in accordance with the Multijurisdictional Disclosure System established between Canada and the United States.
The Company has the right to terminate the LPC Purchase Agreement at any time at no cost or penalty. LPC has agreed not to engage in any short selling or hedging activity of any kind in the Common Shares. As consideration for LPC’s obligation to purchase Common Shares from the Company at its direction under the LPC Purchase Agreement, the Company issued 66,812 Common Shares to LPC as a commitment fee. The LPC Purchase Agreement provides that the Company may not issue or sell any Common Shares to LPC under the LPC Purchase Agreement which, when aggregated with all other Common Shares then beneficially owned by LPC and its affiliates (as calculated pursuant to Section 13(d) of the U.S. Securities Exchange Act of 1934, as amended (the “Exchange Act”), and Rule 13d-3 thereunder), would result in LPC beneficially owning more than 9.99% of the outstanding Common Shares. On July 31, 2023, the Company announced that it had suspended all sales under the LPC Purchase Agreement in connection with the August 2023 Offering (as defined herein). On August 23, 2023, the Company also announced the filing of a prospectus supplement to the Company’s base shelf prospectus dated August 17, 2023, as amended on December 22, 2023, April 8, 2024 and January 6, 2025 (the “2023 Base Shelf Prospectus”), requalifying the Company’s LPC Purchase Agreement on the same terms as those entered into on May 30, 2023 with LPC. On November 9, 2023, the Company announced that it has, again, suspended all sales under the LPC Purchase Agreement.
On June 5, 2023, the Company announced changes to its scientific management team. Following the achievement of the final milestones as contemplated by the terms of the Contribution Agreement, Michael Palfreyman Ph.D. and Brett Greene, who joined the Company following the Adelia Transaction, will leave their roles as Chief R&D Officer and Chief Innovations Officer, respectively, and transition into advisory roles at the Company. Alex Nivorozhkin Ph.D., one of Adelia’s founders, will continue in his role as Chief Scientific Officer of the Company.
On June 27, 2023, the Company announced the appointment of Sanford R. Climan as a strategic advisor.
On June 29, 2023, the Company announced the appointment of Aaron Bartlone as Chief Operating Officer of Helus Pharma., effective July 1, 2023. Mr. Bartlone has served as Chief Operating Officer of Helus Pharma’s U.S. subsidiary, Helus US, since March 2021.
On July 12, 2023, the Company announced that it had commenced the development of a streamlined, scalable version of its EMBARK Training Program, known as EMBARKCT.
On July 26, 2023, the Company announced that it had partnered with Worldwide Clinical Trials, a global, full-service contract research organization with deep expertise managing clinical trials for mental health conditions, including MDD.
On August 4, 2023, the Company completed a public offering (the “August 2023 Offering”) of 638,545 units of the Company (the “August 2023 Units”) at a price of $12.92 per August 2023 Unit for gross proceeds of $8,250 pursuant to a supplement to the Company’s short form base shelf prospectus dated July 5, 2021. Each August 2023 Unit is comprised of one Common Share and one Common Share purchase warrant (the “August 2023 Warrants”). Each August 2023 Warrant is exercisable to acquire one Common Share at a price of $15.20 for a period of 60 months from issuance, subject to acceleration in certain circumstances. The August 2023 Warrants are governed by a warrant indenture dated August 4, 2023, entered into with Odyssey Trust Company, as warrant agent (the “Warrant Indenture”). The August 2023 Offering was completed pursuant to an underwriting agreement among the Company, Cantor Fitzgerald & Co. as the sole book-running manager, and A.G.P./Alliance Global Partners as lead manager. In connection with the August 2023 Offering, the Company paid the underwriters a cash commission of $379 and incurred additional share issuance costs, being professional fees of $465.
On August 15, 2023, the Company announced that the USPTO had granted U.S. patent 11,724,985, to a NSA in the Company’s HLP003, investigational drug program. The patent, which is expected to provide exclusivity until 2041, includes composition of matter claims to deuterated tryptamines in support of the Company’s clinical-stage programs, HLP003, a proprietary NSA, and HLP004, a proprietary NSA, in addition to other of the Company’s pre-clinical programs, as well as claims directed towards methods of treating MDD and treatment-resistant depression.
On August 23, 2023, the Company announced the filing of a prospectus supplement under the 2023 Base Shelf Prospectus to renew its previously established at-the-market equity program (the “2023 ATM Program”) that allowed the Company to issue and sell up to $35,000 of Common Shares from treasury to the public, from time to time. Distributions of Common Shares under the 2023 ATM Program were made pursuant to the terms and conditions of an at-the-market equity distribution agreement (the “2023 Distribution Agreement”) dated August 23, 2023 among the Company, Cantor Fitzgerald Canada Corporation and Cantor Fitzgerald & Co. The 2023 ATM Program was effective until February 10, 2025, when it was terminated in accordance with the terms of the 2023 Distribution Agreement.
On August 28, 2023, the Company entered into the Arrangement Agreement with Small Pharma pursuant to which Helus Pharma Corp. agreed to acquire all of the issued and outstanding shares of Small Pharma (each, a “Small Pharma Share”) in an all-equity business combination transaction to be completed by way the Arrangement.
On September 5, 2023, the Company announced that the USPTO had granted U.S. patent 11,746,088, covering composition of matter for deuterated tryptamine compounds and pharmaceutical compositions thereof, with exclusivity until 2041.
On September 13, 2023, Small Pharma was granted an interim order (the “Interim Order”) by the Supreme Court of British Columbia (the “Court”) regarding the Arrangement. The Interim Order authorized Small Pharma to proceed with various matters relating to the Arrangement, including the holding of a special meeting of Small Pharma shareholders to consider and vote on the Arrangement. Completion of the Arrangement was conditional upon receipt of a final order by the Court. Small Pharma was granted a final order by the Court on October 17, 2023.
On September 26, 2023, the Company announced an agreement with Fluence, a leading continuing education organization in psychedelic therapy, to support the streamlining and scaling of the Company’s EMBARK facilitator training program in preparation for a multi-site, global Phase 3 trial of HLP003, its proprietary NSA in development for the potential treatment of MDD.
On October 12, 2023, the Company held an annual and special meeting of shareholders (the “Special Meeting”) in connection with, among other things, the Arrangement. At the Special Meeting, shareholders of the Company passed an ordinary resolution approving the issuance by the Company of up to such number of Common Shares as may be required to be issued pursuant to the Arrangement in accordance with the terms of the Arrangement Agreement.
On October 23, 2023, the Company completed the Arrangement and issued 0.00634 Common Shares for every one Small Pharma Share outstanding, resulting in a total of 2,130,138 Common Shares being issued to Small Pharma shareholders.
On October 25, 2023, the Company announced that the United States Patent and Trademark Office has issued two patent grants that offer protection for its HLP004 program. These patents are United States patent no. 11,771,681, which provides composition of matter protection for certain deuterated analogs of DMT; and United States patent no. 11,773,062, which provides protection for the medical use and the novel, efficient and scalable synthesis of certain analogs of DMT.
On October 26, 2023, the Company announced that the European Patent Office had granted a patent protecting Helus Pharma Corp’s proprietary NSAs. EP patent no. 4,031,529 provides composition of matter protection for certain deuterated tryptamine compounds, including deuterated NSAs within the HLP003 program and deuterated analogs of DMT within Helus Pharma’s HLP004 program, as well as their medical use.
On October 31, 2023, the Company announced positive Phase 2 interim results for HLP003, its orally delivered deuterated NSA, demonstrating a rapid, robust and statistically significant reduction in symptoms of depression three weeks following a single 12mg dose compared to placebo. At the 3-week primary efficacy endpoint, the reduction in MDD symptoms, defined as change from baseline in MADRS total score, was superior in participants assigned to HLP003 compared to the participants who received placebo by 14.08 points (p=0.0005, Cohen’s d=2.15).
On November 14, 2023, the Company completed a public offering (the “November 2023 Offering”) of 1,754,386 units of the Company (the “November 2023 Units”) at a price of $17.10 per November 2023 Unit for gross proceeds of $30,000 pursuant to a supplement to the Company’s short form base shelf prospectus dated August 17, 2023. Each November 2023 Unit is comprised of one Common Share and one Common Share purchase warrant (the “November 2023 Warrants”). Each November 2023 Warrant is exercisable to acquire one Common Share at a price of $19.38 between May 14, 2024, and May 14, 2029, subject to acceleration in certain circumstances. The November 2023 Offering was completed pursuant to an underwriting agreement between the Company A.G.P./Alliance Global Partners, acting as the sole book-running manager. In connection with the November 2023 Offering, the Company paid the underwriter a cash commission of $1,530 and incurred additional share issuance costs being professional fees of $247.
On November 30, 2023, the Company announced positive Phase 2a topline results for HLP003, showing rapid and robust improvements in symptoms of depression after single doses of HLP003, with an average 14.1 point difference in MADRS score reduction between HLP003 and placebo which was statistically
significant at 3 weeks (p<0.0001). The study also demonstrated a clear incremental benefit of a second dose, with a further 5.8 point improvement on the MADRS total score with a second dose of HLP003 (12 mg) at 6 weeks, and 79% of patients were in remission from depression at 6 weeks after two doses of HLP003 (12 mg). HLP003 exhibited a favorable safety and tolerability profile with no treatment-related serious adverse events at 12 mg and 16 mg doses.
On December 6, 2023, the Company announced that the USPTO had granted U.S. patent 11,834,410 in support of its HLP003 program.
On January 8, 2024, the Company announced positive topline results from its Phase 1 studies of its proprietary deuterated molecules, HLP004 and SPL028. The Phase 1 HLP004 study results showed that IV HLP004 demonstrated robust and rapid-onset pharmacological effects at lower doses compared to the non-deuterated molecule. These pharmacological effects were rapid in onset when administered as an IV bolus over five minutes and persisted for about 40 minutes after the bolus without the need for an extended infusion. The Phase 1 SPL028 study identified an intramuscular (“IM”) dose of SPL028 that resulted in desired pharmacological effects,, with a total duration ranging from 55 to 120 minutes. Both HLP004 (IV) and SPL028 (IM and IV) were well-tolerated with no serious adverse events, and the majority of adverse events were mild to moderate and self-limiting.
On January 23, 2024, the Company announced that it had received FDA clearance to initiate a Phase 2 study of HLP004 in GAD.
On February 7, 2024, the Company announced that the Japan Patent Office has granted JP patents 2023-500532 and 2023-533436. The patents, which are expected to provide exclusivity until at least 2040 and 2041, respectively, include protection for a synthesis method for the preparation of DMT and dDMT and injectable formulations within the Company’s proprietary HLP004 program in clinical development for the treatment of GAD.
On March 13, 2024, the Company announced that the FDA had granted Breakthrough Therapy Designation (“BTD”) to its HLP003 program for the adjunctive treatment of MDD. The BTD provides an expedited review pathway, as well as increased access to FDA guidance on trial design, with the potential to reduce drug development timelines.
On March 14, 2024, the Company announced a positive End-of-Phase 2 meeting with the FDA for HLP003 for the adjunctive treatment of MDD.
On March 13, 2024, the Company reported positive four-month durability data from the Phase 2a study of HLP004 in MDD. These results showed robust, sustained and statistically significant improvements in depression symptoms at four months with two doses of HLP003 (12mg or 16mg). The mean reduction from baseline in the MADRS total score was approximately 22 points from baseline in both dosing cohorts. Additionally, 60% of patients on 12 mg and 75% on 16 mg were in remission from depression following 2 doses (MADRS score </= 10).
On March 15, 2024, the Company announced that it had initiated a Phase 2 study of IM HLP004 in participants with moderate to severe GAD.
On March 19, 2024, the Company completed a private placement (the “March 2024 Offering”) of 9,179,927 Common Shares at a price of $16.34 per Common Share for gross proceeds of $150,000. Pursuant to the terms of the March 2024 Offering, on April 8, 2024, the Company amended the 2023 Base Shelf Prospectus to provide that the securities that may be offered and issued thereunder will include
distributions by various selling securityholders. Further, on April 17, 2024, the Company filed a prospectus supplement to the 2023 Base Shelf Prospectus, in order to qualify the periodic resale of 8,763,941 Common Shares issued to certain non-Canadian investors pursuant to the March 2024 Offering. The March 2024 Offering was completed pursuant to an agency agreement (the “March 2024 Agency Agreement”) among the Company, Bloom Burton Securities Inc. as the lead agent, and Haywood Securities Inc. In connection with the March 2024 Offering, the Company paid the agents a cash commission of $8,665 and incurred additional share issuance costs being professional fees of $371.
Year ended March 31, 2025
On April 1, 2024, pursuant to the provisions of the OBCA, Small Pharma completed a horizontal amalgamation with Helus Pharma Corp., with Helus Pharma Corp. being the resulting entity. As a result of this amalgamation, Cybin UK Ltd. T/A Helus is now a wholly-owned subsidiary of Helus Pharma Corp.
On April 5, 2024, the Company granted options to purchase up to 308,294 Common Shares, of which 134,872 were granted to employees, 144,738 were granted to officers of the Company and 28,684 were granted to consultants. The granted options have an exercise price of CA$21.28 per Common Share. All of the options expire on April 5, 2029. The granted options are subject to different vesting schedules. 38,536 options vested immediately and 269,758 options vest over two years. The aggregate estimated grant date fair value of these options was determined to be $3,397, calculated using the Black-Scholes option pricing model.
On April 8, 2024, the Company amended the 2023 Base Shelf Prospectus to provide that the securities that may be offered and issued thereunder will include distributions by various selling security holders.
On April 16, 2024, the Company announced that the United States Patent and Trademark Office granted U.S. patent 11,958,807 in support of its HLP003 program in MDD.
On April 17, 2024, the Company filed a prospectus supplement to the 2023 Base Shelf Prospectus, in order to qualify the periodic resale of 8,763,941 Common Shares issued to certain non-Canadian investors pursuant to the March 2024 Offering.
On April 18, 2024, the Company announced that its research manuscript, entitled “Synthesis and Structure-Activity Relationships of 2,5-dimethoxy-4-substituted phenethylamines and the discovery of CYB210010: A potent, orally bioavailable and long-acting serotonin 5-HT2 receptor agonist,” was published in the Journal of Medicinal Chemistry, a prestigious bi-weekly peer-reviewed publication.
On May 5, 2024, the Company cancelled options to purchase up to 1,199,655 Common Shares with exercise prices ranging from CA$27.17 to CA$119.70.
On June 11, 2024, the Company announced that Dr. Atul R. Mahableshwarkar M.D., DLFAPA, joined the Company as Senior Vice President, Clinical Development. Dr. Mahableshwarkar will lead the development of the HLP003 program.
On August 13, 2024, the Company announced that it held a Type B Initial Comprehensive Breakthrough Therapy Meeting with the FDA. Further to recent industry discussions around the subject of functional unblinding, the Company plans to implement multiple measures to attempt to mitigate the risk of functional unblinding in its pivotal study program. In addition, the studies will include manual and real time artificial intelligence screening of monitoring sessions to ensure monitor fidelity and patient safety.
On September 19, 2024, the Company announced the appointment of Dr. Tom Macek, PharmD, PhD to lead the Company’s HLP004 program.
On September 19, 2024, the Company announced that it had filed articles of amendment to complete the Consolidation. The Consolidation was effective at the opening of trading on September 19, 2024.
On October 24, 2024, the Company announced that the USPTO had granted U.S. patent 12,122,741 (‘741) with claims to the composition of matter of lead preclinical candidates in the Company’s HLP005 phenethylamines program.
On November 13, 2024, the Company announced that it had initiated PARADIGM, a multinational pivotal Phase 3 program evaluating the efficacy and safety of HLP003 for the adjunctive treatment of MDD.
On November 18, 2024, the Company reported positive Phase 2 data for HLP003, demonstrating 12-month efficacy in treating MDD. At 12 months after two 16 mg doses, 100% of participants were responsive to treatment and 71% of participants were in remission. The mean change from baseline in MADRS was approximately -23 points at 12 months after two 16 mg doses. HLP003 demonstrated an excellent safety and tolerability profile. No new adverse events were reported in the 12-month follow up, including no reports of suicidality.
On January 6, 2025, the Company filed amendment No. 3 to the 2023 Base Shelf Prospectus to increase the aggregate amount of securities that may be offered from time to time under the 2023 Base Shelf Prospectus from CA$400,000 to CA$650,000.
On January 15, 2025, the Company announced the launch of its first strategic partnership agreement with Segal Trials in furtherance of the Company’s multinational pivotal Phase 3 program evaluating HLP003 for the adjunctive treatment of MDD.
On February 10, 2025, the Company launched a new at-the-market equity program (the “2025 ATM Program”) to allow the Company to issue and sell up to $100,000 of Common Shares from treasury to the public. In connection with the 2025 ATM Program, the Company entered into an at-the-market equity distribution agreement (the “2025 Distribution Agreement”) dated February 10, 2025, among the Company, Cantor Fitzgerald Canada Corporation and Cantor Fitzgerald & Co. The 2025 ATM Program is effective until the earlier of the issuance and sale of all of the Common Shares issuable pursuant to the 2025 ATM Program and September 17, 2025, unless earlier terminated in accordance with the terms of the 2025 Distribution Agreement.
Year ended March 31, 2026
On April 21, 2025, the Company announced a strategic partnership with Osmind. Through this partnership, the Company will leverage Osmind’s 800-clinic network, point-of-care software, and real-world data to support the commercial preparation for its clinical-stage pipeline.
On April 23, 2025, the Company announced the addition of CenExel iResearch Atlanta and Cedar Clinical Research to its strategic partnership agreement program bringing the total to 18 clinical sites engaged to advance the Company’s multinational Phase 3 PARADIGM program evaluating HLP003 for the adjunctive treatment of MDD.
On May 8, 2025, the Company announced that the USPTO had granted U.S. patent 12,291,499 in support of its HLP003 program in MDD.
On May 15, 2025, the Company announced that it has engaged Thermo Fisher Scientific to provide U.S.-based manufacturing for the HLP003 program. The production of both drug substance and drug product will be performed at Thermo Fisher’s U.S. pharma services manufacturing sites. The Company is working with Thermo Fisher’s pharma services sites in Florence, South Carolina, for Phase 3 clinical supply and future commercialization, and Cincinnati, Ohio, for Phase 3 capsule production and commercialization.
On June 3, 2025, the Company announced that the USPTO had granted U.S. patent 12,318,477 in support of its HLP004 program in development for the treatment of GAD.
On June 4, 2025, the Company completed the voluntary dissolution of its wholly-owned subsidiaries, Natures Journey Inc. and Serenity Life Sciences Inc. These entities were non-operational prior to their dissolution.
On June 30, 2025, the Company announced that it has entered into a securities purchase agreement, as amended on August 12, 2025 (the “High Trail Securities Purchase Agreement”) with High Trail Special Situations LLC (“High Trail”), pursuant to which the Company agreed to sell and issue to High Trail up to $500,000 aggregate principal amount of unsecured convertible debentures, as amended (the “Convertible Debentures”). The sale and issue of $50,000 principal amount of Convertible Debentures was completed on June 30, 2025 (the “Convertible Debenture Private Placement”). The sale and issue of $450,000 principal amount of Convertible Debentures shall be determined at a future date, upon mutual agreement of the parties. In connection with the offering, the Company and High Trail entered into a customary Registration Rights Agreement (“High Trail Registration Rights Agreement”) pursuant to which the Company agreed to provide certain registration rights to High Trail under the U.S. Securities Act of 1933, as amended.
The Convertible Debentures had a two-year term from the closing date (the “Convertible Debenture Term”). On closing, the Company pre-paid guaranteed interest of $5,500, equal to 11% of the amount issued for the Convertible Debenture Term (the equivalent of 5.5% per annum). During the year ended March 31, 2026, High Trail converted portions of the Convertible Debentures with aggregate principal amounts $29,850 less issuance costs of $35 for which the Company issued 4,584,856 Common Shares at an average conversion price of $6.5106 which represented the VWAP of the Common Shares for the five trading days immediately prior to each conversion. On November 3, 2025, the Company repaid the remaining outstanding balance of the Convertible Debentures. The Company paid a total of $22,765 which included repayment of the remaining principal of $20,150, as well as early repayment fees of $2,615.
On July 17, 2025, the Company announced that it has received approval from the MHRA to commence EMBRACE, the second pivotal study in PARADIGM, the Company’s Phase 3 multinational program evaluating HLP003, a proprietary NSA.
On August 7, 2025, the Company announced that it had received European approval for the EMBRACE study. The Company’s Clinical Trial Application has been approved by the Irish Medicines Board, acting as the reference Member state, to initiate the EMBRACE study in Ireland, Poland, and Greece.
On August 26, 2025, the Company announced that it had received Australian approval for the EMBRACE study. The Company has received approval through the Clinical Trial Notification scheme, obtained clearance from multiple Ethics Committees of the Australian Therapeutics Goods Administration, and the study site Research Governance Offices, thus allowing the commencement of the EMBRACE study in Australia.
On September 2, 2025, the Company announced that, effective September 2, 2025, Doug Drysdale will step down as the Company’s Chief Executive Officer. The Company’s Co-Founder and President, Eric So, was appointed as Interim Chief Executive Officer by the Board.
On September 8, 2025, the Company announced that it had completed enrollment in the Phase 2 study of HLP004 in GAD.
On September 17, 2025, the Company filed a base shelf prospectus dated September 17, 2025, as amended by Amendment No. 1 dated December 19, 2025, (the “2025 Base Shelf Prospectus”), in each of the provinces and territories of Canada. The 2025 Base Shelf Prospectus qualifies for distribution, from time to time during the 25-month period from the date of the 2025 Base Shelf Prospectus, of up to CA$1,700,000 in the aggregate of Common Shares, warrants, units, debt securities and subscription receipts of the Company.
On October 31, 2025, the Company completed a registered direct offering (the “Registered Direct Offering” of 22,277,750 Common Shares and, in lieu of Common Shares to certain investors, 4,605,500 pre-funded common share purchase warrants at a price of $6.51 per security, generating aggregate gross proceeds of approximately $175,010, with total issuance costs of $10,992 allocated between the Common Shares and pre-funded warrants (the “Offered Securities”). The Offered Securities were sold directly to the certain purchasers pursuant to securities purchase agreements each dated October 28, 2025 (each a “2025 Securities Purchase Agreement”), by and between the Company and each purchaser. Jefferies LLC, TD Securities (USA) LLC and Cantor Fitzgerald & Co., as joint lead placement agents, and Bloom Burton Securities Inc. (collectively, the “Placement Agents”), acted as exclusive placement agents for the Company in respect of the Registered Direct Offering pursuant to the terms and conditions of a placement agency agreement dated October 27, 2025 between the Company and the Placement Agents (the “Placement Agency Agreement”). Each pre-funded warrant is exercisable for one Common Share at a nominal exercise price and does not expire. Each Common Share and pre-funded warrant was issued together with 0.35 of one Common Share purchase warrant, with each whole warrant exercisable at $8.14 per Common Share until the earlier of June 30, 2027, 30 days following the public release of topline data from the APPROACH trial of HLP003 in MDD, or 30 days after the Company exercises its acceleration right, which may occur if the Common Shares trade at or above $19.53 for five consecutive trading days. The Company used a portion of the net proceeds of the Registered Direct Offering to repay outstanding convertible debentures and intends to use the remaining funds to advance its HLP003, HLP004, and HLP005 programs, as well as for working capital and general corporate purposes.
On December 30, 2025, the Company established a new at-the-market equity program (the “2026 ATM Program”) that allows the Company to issue and sell up to $100,000 of Common Shares from treasury to the public, from time to time. Distributions of Common Shares under the 2026 ATM Program are made pursuant to the terms and conditions of an at-the-market equity distribution agreement (the “2026 Distribution Agreement”) dated December 30, 2026, among the Company, Cantor Fitzgerald Canada Corporation and Cantor Fitzgerald & Co. The 2026 ATM Program is effective until the earlier of the
issuance and sale of all of the Common Shares issuable pursuant to the 2026 ATM Program and October 17, 2027, unless earlier terminated in accordance with the terms of the 2026 Distribution Agreement.
On January 5, 2026, the Company transferred its U.S. stock exchange listing from NYSE American (previous ticker: CYBN) to Nasdaq under the ticker symbol “HELP”. The Company continues to be listed on Cboe Canada under the same “HELP” ticker symbol. Concurrent with the commencement of trading on Nasdaq, the Company announced it will be doing business as Helus (pronounced “Heal Us”) Pharma. The Company expects to seek approval from shareholders to change its legal name to Helus Pharma Inc. at the Company’s next annual and special meeting of shareholders.
On February 10, 2026, the Company announced the appointment of Michael Cola as CEO. See Subsequent Events.
On February 24, 2026, the Company announced the appointment of Dr. Freda Lewis-Hall, DFAPA, MFPM, to its Board.
In February 2026, the Board created a scientific advisory committee (the “Scientific Advisory Committee”) to: (i) assist the Board in its oversight of the Company’s pharmaceutical research, development, manufacturing, regulatory, licensing and acquisition initiatives; (ii) identify and discuss significant emerging trends and issues in pharmaceutical science, technology and regulation and consider the potential impact of such on the Company; and (iii) assist the Board with its interpretation of scientific and clinical development data and management of the Company with the communication of such data to stakeholders. Dr. Freda Lewis-Hall and Dr. Eric Hoskins serve as members of the Scientific Advisory Committee, with Dr. Lewis-Hall acting as chair.
On March 5, 2026, the Company announced topline results from its Phase 2 study of HLP004 in GAD. Key findings include:
•Clinically meaningful efficacy: Patients that received 20mg HLP004 adjunctive to standard of care therapy achieved mean reduction of 10.4-points (p<0.0001) in the HAM-A from baseline at six weeks.
•Efficacy in difficult to treat population: Study population consisted of moderate-to-severe patients who remained symptomatic despite ongoing antidepressant or anxiolytic therapy.
•Durable remission and robust response over time:
◦At six months, the pooled study population showed 67% responders and 39% remitters.
◦Participants randomized to both 20 mg and 2mg dosing arms experienced meaningful subjective effects and showed clinically significant responses over standard of care, with 59% meeting the criteria for response and 32% for remission in the 20mg arm and a 30% responder and remitter rate in the 2mg arm at week 6.
•Commercially scalable clinic time: Short in-clinic treatment experience with acute drug effects lasting approximately 90 minutes and discharge readiness within approximately three hours3, fitting within the treatment paradigm of existing interventional psychiatry clinics.
•Well tolerated: Favorable tolerability profile with no drug-related serious adverse events or suicidality-related safety signals.
3 In Phase 1 study at 30 mg dose.
Subsequent Events
On April 16, 2026, the Company announced the appointment of Dr. Ken Kramer, PhD as Senior Vice President, Medical Affairs, effective immediately.
On April 20, 2026, the Company announced that Michael Cola stepped down as Chief Executive Officer, effective immediately, at the request of the Board. The Board appointed the Company’s Co-founder and Executive Chairman, Eric So, to resume the role of Interim Chief Executive Officer, effective immediately, while a search for a successor is conducted. Mr. So previously served as Interim Chief Executive Officer and brings continuity of leadership during this transition. In connection with Mr. Cola’s departure, 975,000 RSUs and 325,000 PSUs expired. Mr. Cola’s employment agreement provides for 12 months of severance totaling $750.
On April 23, 2026, the Company announced the addition of Dr. Robert Langer and Dr. Stephen Brannan to its Scientific Advisory Board. Helus Pharma’s Scientific Advisory Board supports the Company’s commitment to advancing its pipeline through disciplined drug development and scientific rigor.
On April 28, 2026, the Company announced a collaboration with TARA Mind to support clinical trial recruitment for its PARADIGM HLP003 Phase 3 program for Major Depressive Disorder (“MDD”), while expanding mental health awareness and access within the veteran community.
On June 24, 2026, the Company announced that enrollment in the APPROACH Phase 3 clinical trial of HLP003 for the adjunctive treatment of MDD is progressing as planned and has surpassed 86% enrollment.
On June 25, 2026, the Company completed an underwritten offering of 10,309,280 Common Shares at an offering price of $4.85 per Common Share, for aggregate gross proceeds of $50,000 (the “2026 June Offering”), pursuant to an underwriting agreement dated June 23, 2026, between the Company and Cantor Fitzgerald & Co., Barclays Capital Inc., Bloom Burton Securities Inc., and Lucid Capital Markets (the “June 2026 Underwriting Agreement”). The Company intends to use the net proceeds from the Offering to progress the Company’s HLP003 for MDD with Phase 3 APPROACH data expected in the fourth quarter of 2026, HLP004 for generalized anxiety disorder, and HLP005 programs, and for working capital and general corporate purposes. In consideration for their services, the Company paid to the underwriters a cash commission of $3,000.
Significant Acquisitions and Dispositions
The Company has not completed any significant acquisitions or dispositions during the fiscal year ended March 31, 2026 for which disclosure is required under Part 8 of NI 51-102.
DESCRIPTION OF THE BUSINESS
Helus Pharma is a clinical-stage pharmaceutical company on a mission to provide treatments designed to foster durable improvements in mental health and help minds heal. Helus Pharma strategically innovates NSAs through rigorous patient-centered research and clinical excellence.4
Helus Pharma’s research and development work focuses on a three-pillar strategy that leverages the Company’s core competencies in preclinical innovation and clinical development. This strategy supports the creation of intellectual property (“IP”) focused on developing the Company’s platform technology to develop NSAs, the progression of clinical development programs studying certain of these NSAs, including HLP003, HLP004, HLP005, and an expansive list of preclinical molecules to facilitate future drug development opportunities. These name changes did not alter the underlying scientific foundations, development plans, or regulatory status of the programs, but were implemented to provide greater consistency, clarity, and alignment with the Helus Pharma brand as the Company advances its pipeline through clinical development and prepares for potential commercialization.
Headquartered in Canada, Helus Pharma is operational in Canada, the United States, the United Kingdom, Poland, Greece, Australia, the Netherlands, and Ireland. For Company updates and to learn more about Helus Pharma, visit www.helus.com or follow the Company on X, LinkedIn, YouTube and Instagram.
Advancement of Mental Healthcare
The Company is conducting research and development of next-generation therapeutics that aim to address unmet needs in the treatment of mental health and neurological conditions. This comprehensive development work is predicated on structural modifications of known tryptamine and phenethylamine derivatives to improve their pharmacokinetic properties while maintaining their respective pharmacology.
Across its extensive research and development programs, Helus Pharma is evaluating a wide array of novel, synthetic active pharmaceutical ingredients (“API”) intended to be delivered through innovative drug delivery systems including via inhalation, via intravenous (“IV”), and intramuscular, or subcutaneous administration.5
The Company intends to apply for regulatory approval for therapies targeting indications such as MDD, AUD, GAD and potentially other various mental health conditions.6 The Company is also developing compounds that may have the potential to address neuroinflammation, central nervous system (“CNS”) disorders, and psychiatric disorders.7
4 This is a forward-looking statement that involves material assumptions by the Company. Drug development involves long lead times, is very expensive and involves many variables of uncertainty. Anticipated timelines regarding drug development and recruitment of patients for participation in clinical trials are dependent on various factors and are based on reasonable assumptions informed by current knowledge and information available to the Company. Such statements are informed by, among other things, eligibility and exclusion criteria for the trial, design of the clinical trial, competition with other companies for clinical sites or patients, perceived risks and benefits of the prescription drug product candidate, the number, availability, location and accessibility of clinical trial sites, regulatory guidelines for developing a drug with safety studies, proof of concept studies, and pivotal studies for new drug application submission and approval, and assumes the success of implementation and results of such studies on timelines indicated as possible by such guidelines, other industry examples, and the Company’s development efforts to date.
5 See footnote 4.
6 A material factor and assumption underlying this forward-looking statement is that the Company will be able to successfully negotiate strategic partnerships.
7 See footnote 6.
Further, over the next 12-month period, the Company will continue to seek to establish strategic partnerships that advance the Company’s scientific research and IP for novel compounds and delivery mechanisms.8 The Company will also continue to sponsor select internal and partner-related clinical trials that advance the understanding of safety and efficacy for various NSAs that target mental health and neurological conditions.9
Stage of Development of Principal Products
Like most life sciences and pharmaceutical companies, the Company’s business is focused on research and development and any future revenue will be dependent on a number of factors, including the outcome of the Company’s clinical trials and the receipt of all necessary regulatory approvals.
In order to establish its business operations, Helus Pharma intends to leverage the extensive professional network of its management to build working partnerships with (i) existing producers of products based in Canada, the United States, the EU and the UK to source the pharmaceutical products the Company intends to develop and distribute under its specific brand, and (ii) to explore options to facilitate the development and distribution and sale of its specific brand of pharmaceutical products.10
Prescription drugs are classified and regulated under the federal Food and Drugs Act (Canada) (the “Canadian FDA”). Labeling, marketing and selling of any prescription drug must comply with the Canadian FDA, including by ensuring that the Company’s products are not packaged or marketed in a manner that is misleading or deceptive to a consumer.
In the United States, foods, drugs and dietary supplements are subject to extensive regulation. The Federal Food, Drug, and Cosmetic Act (the “FFDCA”) and other federal and state statutes and regulations govern, among other things, the research, development, testing, manufacturing, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. The Company must ensure that all promotion and marketing, distribution, and labeling of any pharmaceutical products comply with the U.S. regulations, including the FFDCA and the FDA.
The Company holds a Schedule I manufacturing license from the U.S. Drug Enforcement Administration (“DEA”) for its research lab in the Boston area. The license allows the Company to further become a hub for innovation and drug discovery. With the DEA license, the Company has expanded its internal R&D capabilities to support innovative drug discovery and delivery involving Schedule I compounds.
On March 13, 2024, the Company announced that it had been granted Breakthrough Therapy Designation (the “BTD”) by the FDA in respect of HLP003. The BTD provides an expedited review pathway, as well as increased access to FDA guidance on trial design, which has the potential to significantly reduce drug development timelines. The designation includes all “fast track” program features, as well as more
8 See footnote 6.
9 The material factors and assumptions underlying this forward-looking statement are: (a) that the Company will be able to successfully negotiate strategic partnerships; and (b) all necessary approvals for the studies will be obtained.
10 At this time the Company has not entered into commercial supply agreements and has no control over price or conditions. The Company’s assumption is that it will be able to enter into agreements at such a time when there will be sufficient competition in the market which will render prices reasonable.
intensive FDA guidance and discussion of the HLP003 development program, including planned clinical trials and plans for expediting the manufacturing development strategy.
Non-Revenue Generating Projects11
The Company currently has three significant projects, which have not yet generated revenue:
a.HLP003 Program12
b.HLP004 Program13
c.HLP005 Program14
The Company has developed EMBARK, a psychological support model that integrates leading clinical approaches to promote supportive healing with pharmacological interventions for neuropsychiatry. EMBARK’s six clinical domains (Existential-Spiritual, Mindfulness, Body Aware, Affective-Cognitive, Relational, Keeping Momentum) represent the broad spectrum of ways in which therapeutic benefits may arise in treatment and the equally broad training needed to prepare therapists to support them all. The Company launched its EMBARK training program in June 2021, which prepares facilitators to work within all of these domains, while inviting facilitators to bring in their own therapeutic training and expertise in a flexible, yet structured way. The EMBARK curriculum additionally emphasizes trauma-informed, culturally competent, and ethically rigorous care. On April 12, 2023, the Company announced the launch of EMBARK Open Access, a free online foundational training course for facilitation. EMBARK Open Access is the first and only free massive open online course that offers foundational facilitation training for healthcare professionals and people interested in offering psychological support. On July 12, 2023, the Company announced that it has commenced the development of a streamlined, scalable version of its EMBARK Training Program, known as EMBARKCT, which is designed for individuals with existing knowledge, skills, and experience in facilitation. The EMBARKCT training program is expected to enable the Company to effectively screen, qualify, and train facilitators on a multi-site, international level, to provide support and in-person monitoring for study participants receiving the Company’s investigational therapeutics in larger pivotal trials.
The following is a description of each program, including a description of the Company’s plan for such programs, the status of the objectives related to the Company’s plan for such program and anticipated expenditures to advance the program to the next stage of pipeline development.
The allocation of capital towards the Company’s ongoing projects and programs is largely dependent on the success, or difficulties encountered, in any particular portion of the process and therefore the time involved in completing it; in turn the time and costs associated with completing each step are highly dependent on the incremental results of each step and the results of other programs, and the Company’s need to be flexible to rapidly reallocate capital to projects whose results show the greatest potential. As such, it is difficult for the Company to anticipate the timing and costs associated with taking the projects to their next planned stage, and the Company cannot make assurances that the estimates reflected in this AIF will prove to be accurate, as actual results and future events could differ materially from those anticipated. Accordingly, investors are cautioned not to put undue reliance on the estimates reflected in this AIF.
11 All quarter references in this section are based on calendar year-end.
12 Formerly named the Deuterated Psilocin Program (CYB003)
13 Formerly named the Deuterated Dimethyltryptamine Program (CYB004).
14 Formerly named the Phenethylamine and Tryptamine Derivatives Program (CYB005).
Moreover, identifying the timing and costs of such projects beyond their immediate next steps go to the core differentiating factors with respect to the Company and its competitors. The disclosure of prospective costs and timing other than as already disclosed by the Company would negatively impact shareholder value and undermine the Company’s proprietary technology. In keeping with pharmaceutical industry practice, it is the Company’s policy to disclose these details in conjunction with its financial statements, and to publicly disclose published patent applications, published scientific papers, scientific symposia and the attainment of key milestones only. In addition, the premature disclosure of proprietary data would have a material and adverse effect on the Company’s patent and other intellectual property rights and could result in the breach of confidentiality obligations.
About the HLP003 Program
The Company has been investigating the development of short-acting NSAs with the aim of creating clinical development candidates, utilizing (i) the chemical modification of known serotonergic agonists through the selective substitution of hydrogen atoms with deuterium (i.e. deuteration); and (ii) the combination of such deuterated tryptamine derivative molecules with selected drug delivery methods, including but not limited to oral, inhalation methods, IV and intramuscular delivery.
The Company’s lead program, HLP003, is an orally delivered deuterated NSA that has been granted FDA BTD for the potential adjunctive treatment of MDD. HLP003 aims to address the limitations of first-generation serotonergic agonists, including side effects, scalability and accessibility of treatment.
The Company completed its HLP003 Investigational New Drug (“IND”)-enabling preclinical studies and Chemistry, Manufacturing and Control (“CMC”) development, including the production of clinical materials required for clinical trials, in the second quarter of calendar 2022. In the same period, the Company submitted an IND application to the FDA and received a “may proceed letter” and IND application clearance from the FDA as well as Institutional Review Board (the “IRB”) approval in the U.S. to commence its first-in-human Phase 1/2a study of HLP003 in participants with moderate to severe MDD. The Company had engaged Clinilabs Drug Development Corporation (“Clinilabs”), a full-service contract research organization with deep expertise in central nervous system drug development, to carry out the Phase 1/2a clinical trial of HLP003.
About the Completed HLP003 Phase 1/2a Clinical Trial
The Phase 1/2a trial was a randomized, double-blind, placebo-controlled study evaluating HLP003 in participants with moderate to severe MDD and in healthy volunteers. Per a protocol amendment to the initial Phase 1/2a study design that was announced on February 28, 2023, the study introduced healthy volunteers for the lower (sub-therapeutic) dose cohorts and added a bioequivalence cohort to facilitate the transition to pivotal studies. Healthy volunteers received two administrations (placebo/active and active/active) one week apart, and measures of pharmacological effect were assessed after each dose. Participants with MDD received two administrations (placebo/active and active/active) three weeks apart and response/remission were assessed three weeks after each dose. MDD participants in the trial that were being treated with antidepressants were allowed to remain on their antidepressant medication.
The study investigated the safety, tolerability, pharmacokinetics and pharmacodynamics (“PD”), and pharmacological effect of ascending oral doses of HLP003. In participants with MDD, the trial evaluated rapid onset of antidepressant effect on the day of dosing, using the Montgomery-Asberg Depression Rating Scale (“MADRS”), and evaluated the incremental benefit of a second dose of HLP003 when administered at Week 3. The study included an optional period of assessment to evaluate the durability of
treatment effect out to 12 months. The study is listed on ClinicalTrials.gov under Identifier: NCT05385783.
On August 30, 2022, the Company announced that the first two participants had been dosed in the Phase 1/2a study.
On February 28, 2023, the Company announced positive interim safety and pharmacokinetics and pharmacodynamics data from the Phase 1/2a study of HLP003. Interim findings showed that HLP003 exhibited rapid, short-acting effects, low variability in plasma levels, and achieved desired pharmacological effects at low doses. At the 8 mg and 10 mg dose levels, most of the participants reported robust and meaningful pharmacological effects, confirming a complete mystical experience was achieved. All doses evaluated (single oral doses of HLP003 up to 10 mg) were well-tolerated with no serious adverse events reported.
On July 24, 2023, the Company announced that it had completed dosing in Cohort 5 of the Phase 2a portion of the study with no serious adverse events or other adverse events that may preclude continued dosing, with recruitment underway for Cohort 6. The Phase 2a trial, consisting of completed Cohorts 4 and 5 as of the date of the announcement, evaluated two 12 mg doses of HLP003. On August 2, 2023, the Company announced that it had initiated dosing in Cohort 6, the final cohort of the HLP003 Phase 2a study.
On September 21, 2023, the Company announced that it had completed enrollment in its Phase 2a study of HLP003, its proprietary NSA molecule being developed for the potential treatment of MDD. All participants in the sixth, and final, cohort received at least one dose (placebo or 16 mg of HLP003) with several second doses already administered, and no serious adverse events observed in participants. As of that date, HLP003 demonstrated a favorable safety and tolerability profile at all doses evaluated in the five completed cohorts (1 mg, 3 mg, 8 mg, 10 mg, and 12 mg).
On October 3, 2023, the Company announced that it had completed dosing in Cohort 6 of its Phase 2a study of HLP003. The following doses were evaluated in the six cohorts that comprised the Phase 2a study: 1 mg, 3 mg, 8 mg, 10 mg, 12 mg, and 16 mg. As of that date, HLP003 has been shown to be safe and tolerable at all doses evaluated with no serious adverse events or discontinuations due to adverse events having been observed in the final dose cohort.
On October 31, 2023, the Company announced Phase 2a interim results for HLP003, demonstrating a rapid, robust and statistically significant reduction in symptoms of depression three weeks following a single 12 mg dose compared to placebo, in participants with moderate to severe MDD. At the 3-week primary efficacy endpoint, the reduction in MDD symptoms, defined as change from baseline in the MADRS total score, was superior in participants assigned to HLP003 compared to the participants who received placebo by 14.11 points (p=0.0001, Cohen’s d=2.31).
On November 30, 2023, the Company announced positive Phase 2a topline results for HLP003, showing rapid and robust improvements in symptoms of depression after single doses of HLP003, with an average 14.1 point difference in MADRS score reduction between HLP003 and placebo which was statistically significant at 3 weeks (p<0.0001). The study also demonstrated a clear incremental benefit of a second dose, with a further 5.8 point improvement on the MADRS total score with a second dose of HLP003 (12 mg) at 6 weeks, and 79% of patients were in remission from depression at 6 weeks after two doses of HLP003 (12 mg). HLP003 exhibited a favorable safety and tolerability profile with no treatment-related serious adverse events at 12 mg and 16 mg doses.
On March 13, 2024, the Company announced that the FDA had granted BTD to its HLP003 Program for the potential adjunctive treatment of MDD. The BTD provides an expedited review pathway, as well as increased access to FDA guidance on trial design, with the potential to reduce drug development timelines. On March 13, 2024, the Company also reported positive four-month durability data from the Phase 2a study of HLP003 in MDD. These results showed robust, sustained and statistically significant improvements in depression symptoms at four months with two doses of HLP003 (12 mg or 16 mg):
•Average mean reduction from baseline in the MADRS total score across 2 cohorts was approximately 22 points from baseline in both dosing cohorts.
•60% of patients on 12 mg and 75% on 16 mg were in remission from depression following 2 doses (MADRS score </= 10).
On August 13, 2024, the Company announced that it held a Type B Initial Comprehensive Breakthrough Therapy Meeting with the FDA. Further to recent industry discussions around the subject of functional unblinding, the Company plans to implement multiple measures to attempt to mitigate the risk of functional unblinding in its pivotal study program, as follows:
•the pivotal study program will include a study with a three-arm design with a high dose, mid-dose, and placebo arm. Patients will not know if they received the therapeutic high dose or the sub therapeutic mid-dose, mitigating the unblinding to an extent and addressing potential expectancy bias;
•the studies will utilize remote, independent, blinded raters who will not have any information on the dose received or the participant’s dosing experience;
•the reporting of effects during the dosing session will be fire-walled to ensure that the study team stays blinded;
•the studies will recruit participants who are largely naïve to serotonergic agonists that result in non-ordinary states of consciousness to reduce the impact of expectancy bias; and
•the studies will assess long-term efficacy data points up to one year, to outlast any potential expectancy effects.
In addition, the studies will include manual and real time artificial intelligence screening of monitoring sessions to ensure monitor fidelity and patient safety.
On November 18, 2024, the Company reported positive Phase 2 data for HLP003, demonstrating 12-month efficacy in treating MDD. At 12 months after two 16 mg doses, 100% of participants were responsive to treatment and 71% of participants were in remission. The mean change from baseline in MADRS was approximately -23 points at 12 months after two 16 mg doses. HLP003 demonstrated an excellent safety and tolerability profile. No new adverse events were reported in the 12-month follow up, including no reports of suicidality.
About the Phase 3 PARADIGM Pivotal Program
On November 13, 2024, the Company announced that it had initiated PARADIGM, a multinational pivotal Phase 3 program evaluating HLP003 for the potential adjunctive treatment of MDD.
The Company’s Phase 3 program comprises three pivotal efficacy studies as outlined below. Dosing is underway in APPROACH, the first pivotal study, and patient rollover is ongoing into EXTEND (as defined herein).
Pivotal study 1: APPROACHTM (A Phase III, Placebo-Controlled, Randomized, Double-Blind Trial of Oral Doses of HLP003 to Assess Combined Safety and Efficacy in Humans with Major Depressive Disorder)(“APPROACH”).
•Participants (n=220) will be randomized 1:1 to receive either 16 mg of HLP003 (n=110) or inactive placebo (n=110). Each study arm will evaluate a two-dose regimen, with doses administered three weeks apart. The study will enroll patients suffering from moderate to severe MDD (MADRS≥24) who are on a stable dose of antidepressant medication but are responding inadequately.
•The primary endpoint will be change in depressive symptoms as measured by change in MADRS from baseline at six weeks after the first dose.
APPROACH will enroll participants at approximately 45 clinical sites across the U.S.
Pivotal study 2: EMBRACETM (An Efficacy and Safety, Phase III, Multi-center, Double-Blind, Randomized Controlled Study Comparing 2 Active Doses of HLP003 and Placebo in Eligible Participants With Major Depressive Disorder) (“EMBRACE”).
•Participants (n=330) will be randomized 1:1:1 to receive 16 mg of HLP003 (n=110), 8 mg of HLP003 (n=110), or inactive placebo (n=110). Each arm will evaluate a two-dose regimen, with doses administered three weeks apart. The study will enroll patients suffering from moderate to severe MDD (MADRS≥24) who are on a stable dose of antidepressant medication but are responding inadequately.
•The primary endpoint will be change in depressive symptoms as measured by change in MADRS from baseline at six weeks after the first dose.
EMBRACE is expected to enroll at approximately 60 clinical sites, with minimal site overlap with the APPROACH study.
Pivotal study 3: EXTEND (A Phase III Long-Term Extension Study with Optional Additional Doses of HLP003 to Assess the Safety and Long-term Efficacy in Participants With Major Depressive Disorder) (“EXTEND”).
•Participants from APPROACH and EMBRACE will roll over into EXTEND (up to n=550) after the completion of the 12-week, double-blind, placebo-controlled treatment periods. During EXTEND, all participants who did not respond to treatment in the APPROACH and EMBRACE studies or who relapse during the EXTEND study will be eligible to receive an additional two doses of HLP003 (16 mg) administered three weeks apart. Participants who do not respond to these two doses or relapse again will be eligible to receive an additional single 16 mg dose of HLP003.
Across all three studies, raters will be remote, independent, and blinded with no information on the dose received or the participant’s dosing experience. Effects during the dosing session will be firewalled to ensure that the study team stays blinded.
On January 15, 2025, the Company announced the launch of its first strategic partnership agreement with Segal Trials in furtherance of the Company’s multinational pivotal Phase 3 program evaluating HLP003 for the adjunctive treatment of MDD.
On July 17, 2025, the Company announced that it has received approval from the MHRA to commence EMBRACE, the second pivotal study in PARADIGM, the Company’s Phase 3 multinational program evaluating HLP003, a proprietary NSA.
On August 7, 2025, the Company announced that it had received European approval for the EMBRACE study. The Company’s Clinical Trial Application has been approved by the Irish Medicines Board, acting as the reference Member state, to initiate the EMBRACE study in Ireland, Poland, and Greece.
On August 26, 2025, the Company announced that it had received Australian approval for the EMBRACE study. The Company has received approval through the Clinical Trial Notification scheme, obtained clearance from multiple Ethics Committees of the Australian Therapeutics Goods Administration, and the study site Research Governance Offices, thus allowing the commencement of the EMBRACE study in Australia.
The Company spent approximately $61,797 on the HLP003 Program during the year ended March 31, 2026.
As the Company continues to progress through the HLP003 Program, additional milestones related to its clinical development have been identified. The Company intends to:
•Provide topline efficacy data readout from the first Phase 3 study, APPROACH, in Q4 2026.15
The Company spent approximately $12,766 to initiate enrollment in the second Phase 3 study, EMBRACE, of which approximately $9,105 was spent during the year ended March 31, 2026, and approximately $3,661 was spent in prior fiscal years.
The Company expects to spend approximately $49,85716 to provide topline efficacy data readout from the first Phase 3 study, APPROACH, in Q4 2026. Of this amount, approximately $22,454 was spent during the year ended March 31, 2026, and approximately $3,717 was spent in prior fiscal years, resulting in an
15 There is no assurance that this timeline will be met or that the program will advance to clinical trials, at all. Drug development involves long lead times, is very expensive and involves many variables of uncertainty. Anticipated timelines regarding drug development and recruitment of patients for participation in clinical trials are dependent on various factors and are based on reasonable assumptions informed by current knowledge and information available to the Company. Such statements are informed by, among other things, eligibility and exclusion criteria for the trial, design of the clinical trial, competition with other companies for clinical sites or patients, perceived risks and benefits of the prescription drug product candidate, the number, availability, location and accessibility of clinical trial sites, regulatory guidelines for developing a drug with safety studies, proof of concept studies, and pivotal studies for new drug application submission and approval, and assumes the success of implementation and results of such studies on timelines indicated as possible by such guidelines, other industry examples, and the Company’s development efforts to date. The Company is currently prioritizing the advancement of its HLP003 Program.
16 The Company had previously estimated that its spending to complete this milestone would be $45,881. See footnote 15.
approximate remaining spend as of March 31, 2026, of $23,686 by the milestone completion in Q4 2026.17
The Company intends to continue funding the HLP003 Program, and is targeting a potential U.S. FDA New Drug Application ("NDA") filing in 202818.
The Company intends to complete future clinical trials for this program in the U.S., Europe, the UK, and Australia.
About the HLP004 Program
The Company’s proprietary HLP004 Program is being developed as an intermittent treatment with the potential for less invasive, more convenient and patient-friendly dosing methods for the potential treatment of GAD. A single IM dose is expected to result in desired pharmacological effects lasting an average of 90 minutes.
Helus Pharma has leveraged clinical data from its five completed clinical trials studying first- and second-generation NSAs to inform and optimize the development of the HLP004 Program. To date, Helus Pharma has completed five clinical trials across various molecules demonstrating proof-of-concept in potentially treating depression, supporting the development of its proprietary deuterated NSA, HLP004, for the potential treatment of anxiety disorders, and providing important dosing insights. The Company holds a significant patent portfolio within the HLP004 program, including patents directed to HLP004 and its structural isotopomers.
Key findings from these completed studies are as follows:
•Phase 2a safety and efficacy data for SPL026 (IV N,N-dimethyltryptamine (“DMT”)) in 34 participants with MDD, demonstrating a clinically relevant and statistically significant reduction in depression symptoms at two weeks after dosing (-7.4 point difference in MADRS between SPL026 and placebo). Durable antidepressant response and remission rates were observed at six months. Among participants who had achieved remission within three months with SPL026, 64% sustained remission to six months.
•Phase 1 study evaluating IM SPL026 supporting IM administration for patient-friendly dosing. The study demonstrated that IM delivery of native DMT is well-tolerated and generates a breakthrough experience lasting approximately 45 minutes.
•Phase 1 study evaluating IM SPL028 supporting IM administration for patient-friendly dosing. The completed Phase 1 study of IV/IM SPL028 in healthy volunteers showed that SPL028 is safe and well-tolerated, and demonstrated that IM dosing of SPL028 produced robust pharmacological effects lasting a short duration (average approximately 90 minutes) in the majority of subjects.
•Phase 1b study evaluating the safety and efficacy of SPL026 in conjunction with selective serotonin reuptake inhibitors (“SSRIs”) in 17 participants with MDD, demonstrating no relevant drug-drug interactions, a favorable safety profile and enhanced efficacy when SPL026 was administered with SSRIs, and a 92% remission rate at 4 weeks in the DMT + SSRI combination cohort (n=12).
17 See footnote 15.
18 See footnote 15.
•Phase 1 results for IV HLP004 demonstrated robust and rapid-onset pharmacological effects at lower doses compared to non-deuterated HLP004, suggesting potential as a short-acting, scalable treatment.
Exploratory analysis of the Phase 2a and Phase 1b data for SPL026 also shows significant improvements in symptoms of anxiety, as measured using the State Trait Anxiety Inventory – Trait version (STAI-T), with a 23 point improvement from baseline at the two week endpoint, in the DMT+ SSRI combination group.
The Company is currently advancing HLP004, a deuterated version of DMT, for the potential treatment of GAD. DMT activates the serotonin 5-HT2A receptor, which is believed to mediate the potential therapeutic effects of DMT. In its regular form, DMT is an unstable molecule rapidly metabolized in the body, which significantly reduces its bioavailability. HLP004, as a deuterated molecule, has the potential to overcome the therapeutic limitations of native DMT. To date, HLP004 has demonstrated robust and rapid-onset pharmacological effects at lower doses compared to native DMT, suggesting potential as a short-acting, scalable treatment. Additionally, learnings from Phase 1 studies of IM SPL028 have supported IM administration as a viable dosing method for deuterated DMT, suggesting the potential for HLP004 to offer more convenient and patient-friendly dosing methods.
HLP004 is secured by a U.S. composition of matter patent with protection through 2041. The patent covers a range of deuterated NSAs and protects HLP004 as a putative new chemical entity.
On June 7, 2022, the Company announced it had entered into an agreement to acquire a Phase 1 DMT study (the “Asset Acquisition”) from Entheon Biomedical Corp. (“Entheon”) to accelerate the clinical development path for HLP004. On July 11, 2022, the Company announced that the Asset Acquisition was completed. The Phase 1 study, previously identified as EBRX-101 and now named HLP004-E, was conducted in the Netherlands. Entheon acted as external consultants to the Company for approximately 10 months after the Asset Acquisition.
On January 12, 2023, the Company announced that it has selected GAD as the target indication for its proprietary molecule, HLP004.
About the Phase 1 HLP004-E Study
The Phase 1 trial was a three-part study evaluating the safety, pharmacokinetics, and pharmacodynamics of escalating doses of DMT and HLP004 in healthy volunteers. The three-part study design was established in a protocol amendment to the initial study design, allowing the Company to commence first-in-human dosing of HLP004 sooner than initially planned. The study provided essential safety and dosing optimization data informing the clinical path forward for HLP004. The HLP004-E study was conducted at the Centre for Human Drug Research in the Netherlands and is one of the largest Phase 1 DMT clinical trials to date.
On November 10, 2022, the Company announced that its HLP004-E Phase 1 trial evaluating IV DMT completed dosing for four out of five cohorts and that the Safety Review Committee had confirmed no safety issues.
On February 1, 2023, the Company announced that it had received approval from an independent ethics committee in the Netherlands to initiate first-in-human dosing of HLP004 through a protocol amendment to its ongoing Phase 1 HLP004-E study.
On February 28, 2023, the Company announced a protocol amendment to the initial Phase 1 study design that would allow the Company to initiate first-in-human dosing of HLP004 sooner than initially planned. Per the protocol amendment, Helus Pharma established a three-part study to include Part A (IV DMT infusion), Part B (IV DMT bolus + infusion) and Part C (IV HLP004 bolus + infusion) in healthy volunteers. The Company was able to rely upon completed preclinical data to gain regulatory authorization to add HLP004 to the HLP004-E DMT Study. The Company also announced confirmatory data from Part A, the single ascending dose portion of the HLP004-E study, which assessed a continuous IV DMT infusion. The Part A data showed a dose-proportional increase in exposure and dose-related increase in behavioral measures of subjective pharmacological effects with IV DMT. IV DMT was also well-tolerated with no safety issues and no serious adverse events within the dose range evaluated.
On May 9, 2023, the Company announced that it had completed dosing for the last subject in Part B of the Phase 1 HLP004-E trial.
On May 24, 2023, the Company announced that it had initiated first-in-human dosing of HLP004 in Part C of the Phase 1 HLP004-E trial.
On January 8, 2024, the Company announced positive topline results from its Phase 1 studies of its proprietary molecules, HLP004 and SPL028.
•The Phase 1 HLP004 study results showed that IV HLP004 demonstrated robust and rapid-onset pharmacological effects at lower doses compared to the non-deuterated molecule. These pharmacological effects were rapid in onset when administered as an IV bolus over five minutes and persisted for about 40 minutes after the bolus without the need for an extended infusion.
•The Phase 1 SPL028 study identified an IM dose of SPL028 that resulted in desired pharmacological effects, with a total duration ranging from 55 to 120 minutes.
•Both HLP004 (IV) and SPL028 (IM and IV) were well-tolerated with no serious adverse events, and the majority of adverse events were mild to moderate and self-limiting.
On January 23, 2024, the Company announced that it had received FDA clearance to initiate a Phase 2 study of HLP004 in GAD.
On March 15, 2024, the Company announced that it had initiated a Phase 2 study of IM HLP004 in participants with moderate to severe GAD.
About the Phase 2 HLP004 Study in GAD
The HLP004-002 Phase 2 study was a randomized, double-blind study which evaluated the safety and efficacy of HLP004 in participants with moderate to severe GAD (GAD-7 score ≥10), with concomitant antidepressant/anxiolytic treatment and co-morbid depression allowed. The study recruited 36 participants, who were be randomized in a double-blind manner, into two groups. The first group received two IM doses of HLP004, three weeks apart, while the second group received two low-dose control administrations of sub-therapeutic doses of HLP004. The primary endpoint was a change in the Hamilton Anxiety Rating Scale score from baseline at six weeks following the first dose with an additional efficacy assessment twelve weeks following the first dose. Other endpoints included the HAM-D (Hamilton Depression Rating Scale), safety assessments, MEQ30 (mystical experience Questionnaire) and
EQ-5D-5L (quality of life assessment). Participants were eligible to enroll into an optional follow up for up to a year.
On September 8, 2025, the Company announced that it had completed enrollment in the Phase 2 study of HLP004 in GAD.
On March 5, 2026, the Company announced topline results from its Phase 2 study of HLP004 in GAD. Key findings include:
•Clinically meaningful efficacy: Patients that received 20mg HLP004 adjunctive to SoC therapy achieved mean reduction of 10.4-points (p<0.0001) in the HAM-A from baseline at six weeks.
•Efficacy in difficult to treat population: Study population consisted of moderate-to-severe patients who remained symptomatic despite ongoing antidepressant or anxiolytic therapy.
•Durable remission and robust response over time:
◦At six months, the pooled study population showed 67% responders and 39% remitters.
◦Participants randomized to both 20 mg and 2mg dosing arms experienced meaningful subjective effects and showed clinically significant responses over SoC, with 59% meeting the criteria for response and 32% for remission in the 20mg arm and a 30% responder and remitter rate in the 2mg arm at week 6.
•Commercially scalable clinic time: Short in-clinic treatment experience with acute drug effects lasting approximately 90 minutes and discharge readiness within approximately three hours for 100% of participants19, fitting within the treatment paradigm of existing interventional psychiatry clinics.
•Well tolerated: Favorable tolerability profile with no drug-related serious adverse events or suicidality-related safety signals.
The Company spent approximately $9,952 on the HLP004 Program during the year ended March 31, 2026.
As the Company continues to progress its HLP004 Program, additional milestones related to its clinical development have been identified. The Company intends to:
•Complete the design of the next study by the end of Q3 202620
The Company spent approximately $9,336 to complete the Phase 2 GAD study of which approximately $4,306 was spent during the year ended March 31, 2026 and approximately $5,030 was spent in prior fiscal years.
The Company spent approximately $810 to provide topline data readout from the Phase 2 GAD study, with the full amount incurred in Q1 2026.
The Company expects to spend approximately $150 to complete the design of the next study by the end of Q3 2026.21 No amounts were spent on this milestone during the year ended March 31, 2026.
The Company intends to continue funding the HLP004 Program.
19 In Phase 1 study at 30 mg dose.
20 See footnote 15.
21 See footnote 15.
About the HLP005 Program
The Company’s Phenethylamine and Tryptamine Derivatives Program (HLP005) is focused on the development of therapeutic phenethylamine and tryptamine derivatives. In Q2 2025, the program, which historically focused on phenethylamine derivatives, was expanded to include tryptamine derivatives. Studies that have been conducted with compounds of a similar chemical structure have demonstrated potential for therapeutic use. The HLP005 program builds upon the current understanding of the mechanisms of action of NSAs and aims to identify key polypharmacological combinations capable of mediating therapeutic effects for target indications. Helus Pharma’s proprietary approach to phenethylamines and tryptamines modification with novel chemistry, proprietary formulations and directed delivery systems has yielded a number of novel, IP-protected leads with diverse pharmacology. Several compounds are now being further studied both in vitro and in vivo for selection of the best development candidates based on qualitative assessment of acute pharmacological effects, mechanistic studies, pharmacokinetics and efficacy and safety of chronic dosing. The Company is investigating the efficacy and durability of the lead compounds to determine their potential for the treatment of a range of psychiatric and neurological conditions.22
In order to assess the feasibility and viability of these phenethylamine and tryptamine derivatives entering clinical studies, the Company has and will continue to contract with reputable and licensed third-party vendors to undertake extensive preclinical characterization of target molecules on the Company’s behalf. These activities include, but are not limited to: the synthesis of such molecules as API at laboratory scale, the development and optimization of production processes for such APIs, the development of stable formulations utilizing these APIs, the development and validation of analytical methods for such formulations, the scale up of API production processes beyond laboratory scale to deliver good laboratory practice and Good Manufacturing Practices ("GMP") material suitable for entry into animal and human studies, studies of the stability of such formulations suitable for human studies, the development of Chemistry, Manufacturing and Controls to meet Current Good Manufacturing Practices (“cGMP”).
In addition, utilizing the expertise of selected third parties, the Company intends to oversee the study of the pharmacokinetic profiles of its formulations in a number of animal models and the completion of ADME profiles. Further, the Company’s licensed third party vendors will be responsible for completing a range of additional preclinical programs including, but not limited to, dose-ranging studies in multiple animal species, toxicity studies in multiple animal species, genotoxicity studies, along with neuropharmacological, pulmonary, and cardiovascular profiling, before the final selection of drug candidates for entry into human trials.
The Company intends to complete these studies, and collect further relevant safety and toxicity data, prior to the filing for any IND application with the FDA, a CTA with Health Canada, or other similar application with regulatory bodies in other jurisdictions.
22 This statement is based on the following material factors and assumptions: (a) the Company assumes it will enter into a contract with a licensed third-party vendor to undertake extensive preclinical characterization of target molecules on the Company’s behalf; (b) the Company anticipates to complete a number of animal models and the completion of ADME profiles; (c) the Company assumes to enter into third party agreements in order to complete a range of additional preclinical programs including but not limited to dose-ranging studies in multiple animal species, toxicity studies in multiple animal species, genotoxicity studies, teratogenicity studies, along with neuropharmacological, pulmonary, and cardiovascular profiling before the final selection of drug candidates for entry into human trials; and (d) obtain an IND and/or a CTA to enter into clinical trials. As of the date hereof, it has not yet completed the aforementioned items. Such statements are informed by, among other things, regulatory guidelines for developing a drug with safety studies, proof of concept studies, and pivotal studies for new drug application submission and approval, and assumes the success of implementation and results of such studies on timelines indicated as possible by such guidelines, other industry examples, and the Company’s development efforts to date.
On October 24, 2024, the Company announced that the United States Patent and Trademark Office granted U.S. patent 12,122,741 with claims to the composition of matter of lead preclinical candidates in the Company’s HLP005 program.
The Company spent approximately $792 on its preclinical Phenethylamine and Tryptamine Derivatives Program during the year ended March 31, 2026.
The Company is currently identifying a viable drug development candidate and completing its assessment of the potential path forward for this candidate, including whether it will be developed internally or by way of potential third party partners. The Company anticipates that its Phenethylamine and Tryptamine Derivatives Program may deliver a drug development candidate during the second half of 2027.
The Company expects to spend approximately $2,53523 to deliver a drug development candidate by the end of the second half of 2027.24 Of this amount, approximately $792 was spent during the year ended March 31, 2026 and approximately $691 was spent in prior fiscal years, resulting in an approximate remaining spend as of March 31, 2026, of $1,052 by the milestone completion in the second half of 202725. The Company intends to continue funding the HLP005 program.
Relationships with Third Parties
The Company’s research and development of its pharmaceutical products is conducted by way of licensed partners. The Company also intends to sponsor clinical and other studies at various clinical trial sites.
Clinilabs Drug Development Corporation
On April 21, 2022, the Company announced that it had partnered with Clinilabs, a global, full-service contract research organization with deep expertise in central nervous system drug development, to carry out the Company’s Phase 1/2a clinical trial of HLP003, its proprietary deuterated psilocin program.
Entheon Biomedical Corp.
On July 11, 2022, the Company completed the acquisition of a Phase 1 DMT study from Entheon. As part of the Asset Acquisition, Entheon assigned its rights under the Master Services Agreement between Entheon and Centre For Human Drug Research (“CHDR”) to the Company. The Company now maintains a direct contractual relationship with CHDR to conduct the HLP004-E trial. CHDR is an independent institute in the Netherlands specializing in innovative early-stage clinical drug research.
23 See footnote 15.
24 The Company had previously estimated that this milestone would be achieved by Q4 2026. Anticipated timelines and expenditures associated with delivering a drug development candidate are based on assumptions that management believes to be reasonable, informed by the Company’s current knowledge, experience, and available data. These expectations reflect the activities required to advance a program through key preclinical stages that support candidate selection. In forming these assumptions, management has considered applicable regulatory guidance, industry benchmarks, and the Company’s development progress to date. The achievement of these timelines and expected costs is subject to various risks and uncertainties, including the successful execution and outcomes of development activities, and accordingly, actual results
may differ materially from current expectations. See also footnote 15.
25See footnote 24.
Mindset Pharma Inc.
On September 27, 2022, the Company entered into an agreement, as amended, with Mindset Pharma Inc. (“Mindset”) to acquire an exclusive license to an extensive targeted class of tryptamine-based molecules. The agreement includes an initial license fee payment by Helus Pharma to Mindset of $500 as well as additional clinical development milestone payments of up to $9,500, with the first milestone payment, in the amount of $500, payable upon completion of a Phase 1 clinical trial. At the sole discretion of Helus Pharma, the milestones may be paid in cash or in Common Shares, or a combination thereof, subject to the approval of the Cboe Canada. There is no assurance that the aforementioned milestones will be met. The agreement also contemplates a sales royalty of approximately 2% for all commercialized licensed products within the scope of the agreement, which is customary for drug licensing agreements of this nature.
Worldwide Clinical Trials
On July 26, 2023, the Company announced that it has partnered with Worldwide Clinical Trials, a global, full-service contract research organization with deep expertise managing clinical trials for mental health conditions, including MDD.
Segal Trials
On January 15, 2025, the Company launched its first strategic partnership agreement with Segal Trials in furtherance of Helus Pharma’s multinational pivotal Phase 3 program evaluating HLP003 for the potential adjunctive treatment of MDD. Segal Trials is a privately held company with a network of six research sites throughout South Florida. Segal Trials has extensive experience conducting research trials with an emphasis on psychiatry, neurology, addiction, and studies involving serotonergic compounds.
Osmind
On April 21, 2025, the Company announced a strategic partnership with Osmind, a leading service provider advancing psychiatry through technology, services, and real-world evidence to bring innovative mental health treatments to patients in need. Through this partnership, the Company will leverage Osmind’s 800-clinic network, point-of-care software, and real-world data to support the commercial preparation for its clinical-stage pipeline.
CenExel iResearch Atlanta and Cedar Clinical Research
On April 23, 2025, the Company announced the addition of CenExel iResearch Atlanta and Cedar Clinical Research to its SPA program, bringing the total to 18 clinical sites engaged to advance Helus Pharma’s multinational Phase 3 PARADIGM program evaluating HLP003 for the potential adjunctive treatment of MDD. The APPROACH study is expected to include approximately 45 clinical sites.
Thermo Fisher Scientific
On May 15, 2025, the Company announced that it has engaged Thermo Fisher Scientific to provide U.S.-based manufacturing for the HLP003 Program. The production of both drug substance and drug product will be performed at Thermo Fisher’s U.S. pharma services manufacturing sites. The Company is working with Thermo Fisher’s pharma services sites in Florence, South Carolina, for Phase 3 clinical
supply and future commercialization, and Cincinnati, Ohio, for Phase 3 capsule production and commercialization.
Other Third-Party Partners
The Company has established contractual sources of synthetic GMP and non-GMP raw materials to support its development operations through licensed third-party suppliers located in Canada, the United States, the UK and Europe. Such raw materials are expected to be, in general, readily available and in adequate supply to meet the Company’s need for development quantities, or custom manufactured on the Company’s behalf.26 The prices of research quantities of novel tryptamine compounds are generally higher than commercial supply prices at significantly larger scale and the Company, therefore, expects its supply prices to reduce over time. Development and production of the Company’s proprietary novel compounds is performed under confidential contractual agreements.
The Company has conducted due diligence on each such third party, including but not limited to the review of necessary licences and the regulatory framework enacted in the jurisdiction of operation.
The allocation of capital towards the Company’s ongoing projects and programs is largely dependent on the success, or difficulties encountered, in any particular portion of the process and therefore the time involved in completing it; in turn the time and costs associated with completing each step are highly dependent on the incremental results of each step and the results of other programs, and the Company’s need to be flexible to rapidly reallocate capital to projects whose results show the greatest potential. As such, it is difficult for the Company to anticipate the timing and costs associated with taking the projects to their next planned stage, and the Company cannot make assurances that the foregoing estimates will prove to be accurate, as actual results and future events could differ materially from those anticipated. Accordingly, investors are cautioned not to put undue reliance on the foregoing estimates.
Moreover, identifying the timing and costs of such projects beyond their immediate next steps go to the core differentiating factors with respect to the Company and its competitors. The disclosure of prospective costs and timing other than as already disclosed by the Company would negatively impact shareholder value and undermine the Company’s proprietary technology. In keeping with pharmaceutical industry practice, it is the Company’s policy to disclose these details in conjunction with its financial statements, and to publicly disclose published patent applications, published scientific papers, scientific symposia and the attainment of key milestones only. In addition, the premature disclosure of proprietary data would have a material and adverse effect on the Company’s patent and other intellectual property rights and could result in the breach of confidentiality obligations.
The material factors or assumptions used to develop the estimated costs disclosed above are included in the “Cautionary Note Regarding Forward-Looking Information” section above. The actual amount that the Company spends in connection with each of the intended uses of proceeds will depend on a number of factors, including those listed under “Risk Factors” in this AIF or unforeseen events.
The Company has negative cash flow from operating activities and has historically incurred net losses. To the extent that the Company has negative operating cash flows in future periods, it may need to deploy a portion of its existing working capital to fund such negative cash flows. The Company will be required to raise additional funds through the issuance of additional equity securities, through loan financing, or other
26 At this time the Company has not entered into commercial supply agreements and has no control over price or conditions. The Company has assumed that it will be able to enter into commercial supply agreements at such a time when there will be sufficient competition in the market which will render prices reasonable.
means, such as through partnerships with other companies and research and development reimbursements. There is no assurance that additional capital or other types of financing will be available if needed or that these financings will be on terms at least as favourable to the Company as those previously obtained.
Regulatory Environment
| | | | | | | | |
| Business Segment | Current/Proposed Location of Operation | Summary of Applicable Regulatory Frameworks |
Research, development and commercialization of psychedelic-inspired regulation medicines. | Canada, United Kingdom, United States, European Union/Netherlands, Poland, Greece, Australia | The Canadian and United States federal governments regulate drugs through the CDSA and the CSA, respectively, which place controlled substances in a schedule.(1) The United Kingdom regulates drugs through the MDA (through allocation of classes of risk) and MDR (which places controlled substances in a schedule). In the European Union, clinical trials and medicinal products are regulated pursuant to applicable EU legislation, including the EU Clinical Trials Regulation, together with national controlled substances laws in each member state, including the Dutch Opium Act in the Netherlands, the Misuse of Drugs Acts and Regulations in Ireland, the Act on Counteracting Drug Addiction and related pharmaceutical legislation in Poland, and Law 4139/2013 and related legislation in Greece. Australia regulates therapeutic goods and controlled substances through the Therapeutic Goods Act and the Poisons Standard, with oversight by the Therapeutic Goods Administration.
Under the CDSA, certain serotonergic agonists, including certain NSAs are currently a Schedule III drug.(2)
Under the CSA, certain serotonergic agonists, including certain NSAs are currently a Schedule I drug.(3)
Under the MDA and MDR, certain serotonergic agonists, including certain NSAs are currently classified as Class A drugs and Schedule 1 drugs, respectively.(4)
In the European Union and Australia, certain serotonergic agonists, including certain NSAs, are regulated as controlled or prohibited substances under applicable national scheduling regimes, subject to strict licensing and authorization requirements for research and clinical use.(5)
Under the Dutch Opium Act, DMT is classified in the Netherlands as a List 1 Drug (6) |
Notes:
(1) In both Canada and the United States, the applicable federal government is responsible for regulating, among other things, the approval, import, sale and marketing of drugs, including serotonergic agonists, whether natural or novel. Health Canada and the FDA have not approved HLP003 or HLP004 as a drug for any indication. It is illegal to possess such substances without a prescription. See “Regulatory Environment – Research and Development”.
(2) For further information on the Canadian regulatory framework, see “Regulatory Environment – Canada”.
(3) For further information on the United States regulatory framework, see “Regulatory Environment – United States”.
(4) For further information on the United Kingdom regulatory framework, see “Regulatory Environment – United Kingdom”.
(5) For further information on the European Union regulatory framework, including EU‑level clinical trial regulation and national controlled‑substances regimes applicable in EU Member States in which the Company operates (including Ireland, Poland and Greece), see “Regulatory Environment – European Union/Netherlands”, “Regulatory Environment – Ireland”, “Regulatory Environment – Poland”, “Regulatory Environment – Greece” and “Regulatory Environment – Australia”.
(6) For further information on the Netherlands regulatory framework, see “Regulatory Environment – Europe/Netherlands”.
Research and Development
The Company is focused on development of next-generation therapies, through research and development of novel chemical compounds and delivery mechanisms and study of such compounds in clinical environments around the world. The Company anticipates growing its pipeline of pharmaceutical products through its internal research, development, proprietary discovery programs, mergers and acquisitions, joint ventures and collaborative development agreements. For the time being, the Company maintains intellectual property generated by its R&D programs through patent filings and as trade secrets. The Company anticipates that as these programs mature more patent applications will be filed and more details about these programs will be disclosed at such time.
NSAs are a class of drug designed to activate serotonin receptors and pathways to improve brain and body function, with some NSAs causing thought, visual and auditory changes, and altered states of consciousness. The pharmacokinetics, pharmacology and human metabolism of the non-deuterated form of HLP003 are well known and well characterized. Once ingested, HLP003 acts on serotonin receptors in the brain to produce its intended effects.
The Company’s research and development must be conducted in strict compliance with the regulations of federal, state, local and regulatory agencies in the United States, European Union, the UK, Australia, Poland, Greece, Ireland and Canada, and the equivalent regulatory agencies in any other jurisdictions in which the Company operates. These regulatory authorities regulate, among other things, the research, manufacture, promotion and distribution of drugs in specific jurisdictions under applicable laws and regulations.
Regulatory Framework
United States
The FDA and other federal, state, local and foreign regulatory agencies impose substantial requirements upon the clinical development, clinical testing, approval, labeling, manufacture, marketing and distribution of drug products. These agencies regulate, among other things, research and development activities and the testing, approval, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, advertising and promotion of any prescription drug product candidates or commercial products. The regulatory approval process is generally lengthy and expensive, with no guarantee of a positive result. Moreover, failure to comply with applicable FDA or other requirements may result in civil or criminal penalties, recall or seizure of products, injunctive relief including partial or total suspension of production, or withdrawal of a product from the market. Anticipated timelines related to regulatory filings are based on reasonable assumptions informed by current knowledge and information available to the Company.
Certain serotonergic agonists, including certain NSAs, are strictly controlled under the federal CSA as Schedule I substances. Schedule I substances by definition have no currently accepted medical use in the United States, a lack of accepted safety for use under medical supervision, and a high potential for abuse. Schedule I and II drugs are subject to the strictest controls under the CSA, including manufacturing and procurement quotas, security requirements and criteria for importation. Anyone wishing to conduct research on substances listed in Schedule I under the CSA must register with the DEA and obtain DEA approval of the research proposal. A majority of state laws in the United States also classify certain serotonergic agonists, including certain NSAs, as Schedule I controlled substances. For any product
containing any Schedule I substance to be available for commercial marketing in the United States, such substance must be rescheduled, or the product itself must be scheduled, by the DEA to Schedule II, III, IV or V. Scheduling determinations by the DEA are dependent on FDA approval of a substance or a specific formulation of a substance.
Research and Development
Because HLP003 and HLP004 are treated as Schedule I substances under the CSA, for any product containing HLP003 or HLP004, or any Schedule I substance to be available for commercial marketing in the United States, such substance must be rescheduled, or the product itself must be scheduled, by the DEA to Schedule II, III, IV or V.
The process required before a prescription drug product candidate may be marketed in the United States generally involves:
•completion of extensive nonclinical laboratory tests, animal studies and formulation studies, all performed in accordance with the FDA’s GLP, Good Clinical and/or GMP regulations;
•submission to the FDA of an IND Application, which the FDA must approve before human clinical trials may begin;
•approval by an IRB or independent ethics committee at each clinical trial site before each trial may be initiated;
•for nearly all new pharmaceutical products, performance of adequate and well-controlled human clinical trials in accordance with the FDA’s regulations, including Good Clinical Practices, to establish the safety and efficacy of the prescription drug product candidate for each proposed indication;
•submission to the FDA of an NDA;
•satisfactory completion of one or more FDA pre-approval inspections of the manufacturing facility or facilities where the drug will be produced to assess compliance with cGMP requirements to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality, and purity; and
•FDA review and approval of the NDA prior to any commercial marketing, sale or shipment of the drug.
The testing and approval process requires substantial time, effort and financial resources, and the Company cannot be certain that the DEA will schedule or reschedule any Schedule I substance or product candidate to Schedule II, III, IV or V, or that approvals for its prescription drug product candidates will be granted on a timely basis, if at all.
Non-clinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animals and other animal studies or through FDA-accepted alternative methods (New Approach Methodologies or NAMs). The results of non-clinical tests, together with manufacturing information and analytical data, are submitted as part of an IND to the FDA. Some non-clinical testing may continue even after an IND is submitted. The IND also includes one or more protocols for the initial clinical trial or trials and an investigator’s brochure. An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions relating to the proposed clinical trials as outlined in the IND and places the clinical trial on a clinical hold. In such cases, the IND sponsor and the FDA must resolve any outstanding concerns or questions before any clinical trials can begin. Clinical trial holds also may be imposed at any time before or during studies due to safety concerns or non-compliance with regulatory requirements.
An IRB board, at each of the clinical centers proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center. An IRB board considers, among other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated benefits. The IRB board also approves the consent form signed by the trial participants and must monitor the study until completed. The FDA, the independent IRB, or the sponsor may suspend or discontinue a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk. There also are requirements governing the reporting of ongoing clinical trials and completed clinical trials to public registries.
The FDA offers a number of regulatory mechanisms that provide expedited or accelerated approval procedures for selected drugs and indications which are designed to address unmet medical needs in the treatment of serious or life-threatening diseases or conditions. These include programs such as BTDs, Fast Track designations, Priority Review and Accelerated Approval, which the Company may need to rely upon in order to receive timely approval or to be competitive.
The Company may plan to seek orphan drug designation for certain indications qualified for such designation. The U.S., E.U. and other jurisdictions may grant orphan drug designation to drugs intended to treat a “rare disease or condition,” which, in the U.S., is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or 200,000 or more individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug available in the United States for this type of disease or condition will be recovered from sales of the product. In the E.U., orphan drug designation can be granted if: the disease is life threatening or chronically debilitating and affects no more than 50 in 100,000 persons in the E.U.; without incentive it is unlikely that the drug would generate sufficient return to justify the necessary investment; and no satisfactory method of treatment for the condition exists or, if it does, the new drug will provide a significant benefit to those affected by the condition. Orphan drug designation must be requested before submitting an NDA. If a product that has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and 10 years in the E.U. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for different indications. After orphan drug designation is granted, the identity of the therapeutic agent and its potential orphan use are publicly disclosed. Orphan drug designation does not convey an advantage in, or shorten the duration of, the development, review and approval process. However, this designation provides an exemption from marketing and authorization fees.
Drugs manufactured or distributed pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, reporting of adverse experiences with the product, and complying with promotion and advertising requirements. The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-market testing, including phase IV clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. In addition, drug manufacturers and their subcontractors involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including current Good Manufacturing Practices, which impose certain procedural and documentation requirements. Failure to comply with statutory and regulatory requirements may subject a manufacturer to legal or regulatory
action, such as warning letters, suspension of manufacturing, product seizures, injunctions, civil penalties or criminal prosecution. There is also a continuing, annual prescription drug product program user fee.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information, requirements for post-market studies or clinical trials to assess new safety risks, or imposition of distribution or other restrictions under a risk evaluation and mitigation strategy.
In the United States, pharmaceutical manufacturers are subject to complex laws and regulations pertaining to healthcare “fraud and abuse,” including, but not limited to, the Anti-Kickback Statute, the federal False Claims Act (the “FCA”), and other state and federal laws and regulations. The Anti-Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase, order, or prescription of a particular drug, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid.
The FCA prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to or approval by the federal government or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. Violations of the FCA can result in very significant monetary penalties and treble damages. The federal government is using the FCA, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the FCA in addition to individual criminal convictions under applicable criminal statutes. In addition, the federal civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
There are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. In addition, a similar federal requirement Section 6002 of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or the Affordable Care Act, commonly referred to as the “Physician Payments Sunshine Act” requires applicable manufacturers to track and report to the federal government certain payments and “transfers of value” made to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members, made in the previous calendar year. There are a number of states that have various types of additional reporting requirements.
Controlled Substances
The CSA and its implementing regulations establish a “closed system” of regulations for controlled substances. The CSA imposes registration, security, recordkeeping and reporting, storage, manufacturing,
distribution, importation and other requirements under the oversight of the DEA. The DEA is responsible for regulating controlled substances, and requires those individuals or entities that manufacture, import, export, distribute, research, or dispense controlled substances to comply with the regulatory requirements in order to prevent the diversion of controlled substances to illicit channels of commerce.
The DEA categorizes controlled substances into one of five schedules — Schedule I, II, III, IV or V — with varying qualifications for listing in each schedule. Schedule I substances by definition have a high potential for abuse, have no currently accepted medical use in treatment in the United States and lack accepted safety for use under medical supervision. For any product containing a Schedule I substance to be available for commercial marketing in the United States, such substance must be rescheduled, or the product itself must be scheduled, by the DEA to Schedule II, III, IV or V. Scheduling determinations by the DEA are dependent on FDA approval of a substance or a specific formulation of a substance.
Facilities that research, manufacture, distribute, import or export any controlled substance must register annually with the DEA. The DEA registration is specific to the particular location, activity(ies) and controlled substance schedule(s). For example, separate registrations are needed for import and manufacturing, and each registration will specify which schedules of controlled substances are authorized.
The DEA may inspect all research and manufacturing facilities to review security, recordkeeping, reporting and handling prior to issuing a controlled substance registration and periodically to ensure continued compliance. The specific security requirements vary by the type of business activity and the schedule and quantity of controlled substances handled. The most stringent requirements apply to researchers and manufacturers of Schedule I and Schedule II substances. Required security measures commonly include background checks on employees and physical control of controlled substances through storage in approved vaults, safes and cages, and through use of alarm systems and surveillance cameras. Once registered, manufacturing facilities must maintain records documenting the manufacture, receipt and distribution of all controlled substances. Manufacturers must submit periodic reports to the DEA of the distribution of Schedule I and II controlled substances, Schedule III narcotic substances, and other designated substances. Registrants must also report any controlled substance thefts or significant losses, and must obtain authorization to destroy or dispose of controlled substances. Imports of Schedule I and II controlled substances for commercial purposes are generally restricted to substances not already available from a domestic supplier or where there is not adequate competition among domestic suppliers. In addition to an importer or exporter registration, importers and exporters must obtain a permit for every import or export of a Schedule I and II substance or Schedule III, IV and V narcotic, and submit import or export declarations for Schedule III, IV and V non-narcotics.
For drugs manufactured in the United States, the DEA establishes annually an aggregate quota for the amount of substances within Schedules I and II that may be manufactured or produced in the United States based on the DEA’s estimate of the quantity needed to meet legitimate medical, scientific, research and industrial needs. The quotas apply equally to the manufacturing of the active pharmaceutical ingredient and production of dosage forms. The DEA may adjust aggregate production quotas a few times per year, and individual manufacturing or procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments for individual companies.
Individual U.S. states also establish and maintain separate controlled substance laws and regulations, including licensing, recordkeeping, security, distribution, and dispensing requirements. A majority of state laws in the United States also treat HLP003 and HLP004 as Schedule I controlled substances. State authorities, including boards of pharmacy, regulate use of controlled substances in each state, including state specific controlled substance registration requirements. Failure to obtain applicable registrations or maintain compliance with applicable requirements, particularly as manifested in the loss or diversion of
controlled substances, can result in enforcement action that could have a material adverse effect on the Company’s business, operations and financial condition. The DEA and/or state regulatory agencies may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to revoke those registrations. In certain circumstances, violations could lead to criminal prosecution.
European Union/Netherlands
The International Narcotics Control Board (“INCB”), a UN entity, monitors enforcement of restrictions on controlled substances. The INCB’s authority is defined by three international UN treaties – the UN Single Convention on Narcotic Drugs of 1961, the UN Psychotropic Convention of 1971 (referred to herein as the UN71), and the UN Convention Against Illicit Traffic in Narcotic Drugs and Psychotropic Substances of 1988, which contains provisions related to the control of controlled substance precursors. EU Member States, including the Netherlands, that have agreed to abide by the provisions of these treaties, each create responsible agencies and enact laws or regulations to implement the requirements of these conventions.
Specific EU legislation establishing different classes of controlled substances is limited to EU regulations that define classes of precursors, or substances used in the illicit manufacture of controlled substances, including Regulation (EC) No. 273/2004 of the European Parliament and the Council of February 11, 2004 and the Council Regulation (EC) No. 111/2005 of December 22, 2004. While EU legislation does not establish different classes of narcotic drugs or psychotropic substances, the Council Decision 2005/387/JHA of May 10, 2005 (which applies only in certain limited situations)27 can provoke a Council Decision requiring EU member states to put a drug under national controls equivalent to those of the INCB. HLP004 is currently classified as a Schedule I substance under the UN71; the EU member states that are party to the UN71, including the Netherlands, have agreed to the following in respect of Schedule I substances:
•prohibit all use except for scientific and very limited medical purposes by duly authorized persons, in medical or scientific establishments which are directly under the control of their Governments or specifically approved by them;
•require that manufacture, trade, distribution and possession be under a special licence or prior authorization;
•provide for close supervision of the activities and acts mentioned in paragraphs (a) and (b);
•restrict the amount supplied to a duly authorized person to the quantity required for his authorized purpose;
•require that persons performing medical or scientific functions keep records concerning the acquisition of the substances and the details of their use, such records to be preserved for at least two years after the last use recorded therein; and
•prohibit export and import except when both the exporter and importer are the competent authorities or agencies of the exporting and importing country or region, respectively, or other persons or enterprises which are specifically authorized by the competent authorities of their country or region for the purpose.
27 Decision 2005/387/JHA was repealed (by Directive (EU) 2017/2103 of the European Parliament and of the Council of November 15, 2017 amending Council Framework Decision 2004/757/JHA in order to include new psychoactive substances in the definition of ‘drug’ and repealing Council Decision 2005/387/JHA), but still continues to apply to new psychoactive substances in respect of which a Joint Report (as referred to in Article 5 of that Decision) has been submitted before November 23, 2018.
As classification of controlled substances may vary among different EU member states, sponsors must be aware of the prevailing legislation in each country where a clinical trial may be conducted. Prior to operating or conducting any preclinical or clinical studies in any other EU member state, Helus Pharma will investigate the specific regulatory requirements of such EU member state. As referenced above, a licence is required for individuals and entities who wish to produce, dispense, import, or export Schedule I substances, but the specific requirements vary from country to country. Currently, DMT is classified in the Netherlands as a List 1 Drug under the Dutch Opium Act (Opiumwet) (the “Dutch Opium Act”) and as such, subject to express authorization being obtained, the production, trade and possession of DMT are prohibited.
In addition to the Dutch Opium Act, two other Dutch Acts may be relevant when it comes to drugs: the Medicines Act and the Commodities Act.
The specific regulatory processes and approvals required may vary among different EU member states and are set forth in the respective legislation of each country. For the Netherlands, there are specific regulatory requirements for the approval of clinical trials that need to be met. Firstly, a CTA (Clinical Trial Application) dossier containing the preclinical and any clinical information along with the proposed clinical trial design must be submitted to an accredited Ethics Committee and to the Central Commission on Research in Humans (the “CCMO”), which is also known as the Competent Authority in The Netherlands. In Dutch, the CCMO is called the ‘Centrale Commissie Mensgebonden Onderzoek’. In cases where the study involves a substance subject to the Dutch Opium Act (such as DMT), an official exemption by Farmatec is needed, which needs to be included in the CTA.
Specific rules for the submission, assessment and conduct of clinical trials with medicinal products are set out in, among others, the EU Clinical Trial Regulation 536/2014 (CTR), which is applicable in the EU as of January 31, 2022 and the Medical Research (Human Subjects) Act (Wet medisch-wetenschappelijk onderzoek met mensen).
On April 26, 2023, the European Commission introduced a comprehensive “pharmaceutical package” aimed at revising the EU’s pharmaceutical legislation. This package includes proposals for a new directive and regulation designed to enhance the availability, accessibility, and affordability of medicines. Additionally, it seeks to boost the competitiveness and attractiveness of the EU pharmaceutical industry while imposing higher environmental standards. The European Parliament has recently looked at these proposals to renovate the EU pharmaceutical legislation, and a newly elected Parliament will take up the proposal following the European elections of June 6-9, 2024.
Research and Development
Regulation (EU) No 536/2014 on Clinical Trials on Medicinal Products for Human Use ( the “CTR”) is applicable as of January 31, 2022, harmonizing the laws, regulations and administrative provisions of the EU Member States relating to the implementation of Good Clinical Practice in the conduct of clinical trials on medicinal products for human use.28 The CTR repealed the Clinical Trials Directive, which had previously been transformed into the respective national laws of Member States. The CTR now applies directly in all Member States without transposition being necessary. Pursuant to the CTR, as of January 31, 2023 sponsors are obliged to use the Clinical Trials Information System (“CTIS”) for regularity submission, authorization and supervision of clinical trials in the EU and the EEA. CTIS thus serves as
28 The CTR does not apply in the UK and UK law on clinical trials is currently based on old EU law (the Clinical Trials Directive), transposed into UK law via the Medicines for Human Use (Clinical Trials) Regulations 2004. An overhaul of UK law on clinical trials has been on-going for a few years, and new legislation on clinical trials in the UK, the Medicines for Human Use (Clinical Trials) (Amendment) Regulations 2024, has been signed into law and is due to come into effect from mid-April 2026.
the single-entry point for submissions by sponsors and for regulatory assessment. In addition to this obligation, sponsors have transferred any ongoing (approved) trials under the CTR to CTIS by January 2025. Further, EMA adopted on October 5, 2023, the “Revised CTIS Transparency Rules” on publishing information about clinical trials submitted through CTIS. To increase transparency, EMA removed the deferral mechanism which allowed sponsors to delay certain data and document publication for up to seven years after the end of their trial. Annex I of the revised rules outlines the timing of information publication for each category of clinical trial and patient population. These new rules became applicable on June 18, 2024, the same day of the launch of the new CTIS portal. In order to smoothen the process of transitioning clinical trials from the Clinical Trial Directive to the CTR, a non-binding guide named “Guidance for the Transition of clinical trials from the Clinical Trials Directive to the Clinical Trials Regulation” (version 4) dated May 2024 is published.
CTIS and the practical aspects thereof are also discussed and explained (among other relevant topics relating to clinical trials) in a quick guide on the rules and procedures of the EU Clinical Trials Regulation called “Clinical Trials Regulation (EU) 536/2014 in practice”, which is published by the Clinical Trials Coordination and Advisory Group (“CTAG”) on December 8, 2023. The current version is dated March, 2024. The objective of the rules is to provide sponsors and investigators a quick guide on the rules and procedures of the CTR with a view to facilitating implementation. In addition to the quick guide, CTAG also published a non-binding Questions & Answers (Version 7.1) that should be read in conjunction with the quick guide and with the European Medicines Agency’s (“EMA”) “Clinical Trials Information System (CTIS): online training modules” in order to gain a better understanding of the legislative changes that are effected by the CTR.
The Investigational Medicinal Product Dossier (“IMPD”) is one of several regulatory documents required for conducting a clinical trial of a pharmacologically API intended for one or more EU Member States. The IMPD includes summaries of information related to the quality, manufacture and control of any IMP (including reference product and placebo), and data from non-clinical and clinical studies. Guidance concerning IMPDs is based on the CTR and on EMA guidelines, some of which were originally developed under the former Clinical Trials Directive but have since been updated to reflect the requirements of the CTR.
The content of the IMPD may be adapted to the existing level of knowledge and the product’s phase of development. When applying for a clinical trial authorization, a full IMPD is required when little or no information about an API has been previously submitted to competent authorities, when it is not possible to cross-refer to data submitted by another sponsor and/or when there is no authorization for sale in the EU. However, a simplified IMPD may be submitted if information has been assessed previously as part of a Marketing Authorization or a clinical trial to that competent authority. Although the format is not obligatory, the components of an IMPD largely resemble the types of information required in clinical trial applications in Canada and the United States. The IMPD need not be a large document as the amount of information to be contained in the dossier is dependent on various factors such as product type, indication, development phase etc.
The assessment of an IMPD is focused on patient safety and any risks associated with the IMP. Whenever any potential new risks are identified the IMPD must be amended to reflect the changes. Certain amendments are considered substantial in which case the competent authority must be informed of the substantial amendment. This may be the case for changes in IMP impurities, microbial contamination, viral safety, transmissible spongiform encephalopathies (e.g. mad cow disease) and in some particular cases to stability when toxic degradation products may be generated.
With the completion of the Asset Acquisition, the Company has an ongoing phase I study to obtain preliminary evidence of the safety and efficacy of infused DMT. Prior to the Asset Acquisition, an investigator’s brochure (including prior safety, preclinical and clinical data), and an IMPD document that includes CMC information and a clinical study protocol and supporting information had been prepared. Approval by the Dutch ethics committee of the Phase 1 Study, planned to be conducted by CHDR will be based on the vast amount of published human and animal studies of DMT. Prior to the Asset Acquisition, preclinical data was not provided as part of the application package; however, limited additional in vivo and in vitro data to support the rationale for human dosing and safety had been included. CHDR and its partner GMP-licensed pharmacy that will be involved in the Phase 1 Study, the Leiden University Medical Center, have all the required approvals to possess and handle DMT for the Phase 1 Study.
Failure of the Company to receive the necessary regulatory approvals required to conduct the Phase 1 Study would have an adverse impact on its business plans and financial condition for a number of reasons including, without limitation: (i) it would cause delays in the Company’s research and development plans; (ii) it may require the Company to expend additional financial and human resources on revising its application package or creating a new one; or (iii) it may require the Company to approach an entirely different regulatory authority in a new jurisdiction, in which case the Company would have to expend a substantial amount of capital and other resources on engaging the appropriate research and development partners and creating an application package that complies with the regulations of that new jurisdiction. Additionally, the Company would be required to spend capital on transferring the DMT materials to the new jurisdiction. All of the foregoing would likely have a negative impact on the Company’s business and financial condition.
Pharmaceutical Products
In accordance with the Dutch Medicines Act (Geneesmiddelenwet), “medicinal products” are defined as: a substance or a combination of substances that is intended to be administered or used for, or is presented in any way as being suitable for, use: (i) the cure or prevention of any disease, defect, wound or pain in human beings, (ii) the making of a medical diagnosis in human beings, or (iii) restoring, improving or otherwise modifying physiological functions in humans by exerting a pharmacological, immunological or metabolic effect.
If a product constitutes a medicinal product, a marketing authorization for the product is required before the product may be placed on the market in the Netherlands. In the EU, marketing authorizations may be obtained through the Centralized procedure, the Decentralized or Mutual Recognition procedures and/or the national procedure. The Centralized procedure is compulsory for medicines intended to treat i.e. cancer, AIDS, neurodegenerative diseases and diabetes and optional for medicines comprising of new active substances not previously approved for the EEA. When applying for a marketing authorization through the Centralized procedure, applications are submitted with the EMA. Where the Centralized procedure is not available but a medicinal product is intended for several EU/EEA Member States, an application for a marketing authorization may be submitted with the competent authority of a single EU/EEA Member State in accordance with the Decentralized procedure. When the assessment of the application results in a decision to grant the marketing authorization, this decision will be recognized by the competent authorities of the other Member States for which the marketing authorization is applied. The Mutual Recognition procedure is similar to the Decentralized procedure, but applies to applications where the medicinal product is already the subject of a marketing authorization in another EU/EEA Member State. Finally, should a medicinal product be intended for the Netherlands only, then the national procedure may be followed by submitting an application with the Dutch Medicines Evaluation Board. It may be remarked that the national procedure is unavailable in case the Centralized procedure is
compulsory or in case an applicant has already submitted an application for and/or obtained a marketing authorization in another Member State. In that case, applications must follow the Mutual Recognition procedure instead.
Companies that manufacture or trade in medicinal products and/or active pharmaceutical ingredients in the Netherlands require a manufacturing authorization or a wholesale distribution authorization. A manufacturing authorization is required for the preparation, trading in, import and export of medicinal products and/or active substances. Here, ‘preparation’ means the total or partial manufacture of medicinal products and/or active substances or the packaging or labelling thereof. ‘Importing’ means the import of medicinal products or active substances from a country outside the EEA into the Dutch territory, while ‘exporting’ means the export of medicinal products or active substances from the Dutch territory to a country outside the EEA. A wholesale distribution authorization is required for one or more activities within the wholesale business, such as procuring, holding, supplying, delivering or exporting medicinal products or active substances which are prepared or imported by a third party. It may be noted that holders of wholesale distribution authorization, other than holders of marketing authorizations, are not authorized to import medicinal products from countries outside the EEA.
Only a natural or legal person established in the Netherlands may obtain either a Dutch marketing authorization or a wholesale distribution authorization. These authorizations concern national permits, meaning that these authorizations are not automatically valid in other EU Member States. Furthermore, in the Netherlands applicants of marketing authorizations and wholesale distributions authorizations must be registered with Farmatec and comply with GDP norms.
Marketing Authorization Regulatory Process
Under the Centralized procedure, pharmaceutical companies submit a single marketing authorization application to the EMA, which provides the basis of a legally binding recommendation that will be provided by the EMA to the European Commission, the authorizing body for all centrally authorized products. This allows the marketing-authorization holder to market the medicine and make it available to patients and healthcare professionals throughout the EU on the basis of a single marketing authorization. EMA’s Committee for Medicinal products for Human Use or Committee for Medicinal Products for Veterinary Use carry out a scientific assessment of the application and give a recommendation on whether the medicine should be marketed or not, under any particular dosing regime. Although, under EU law, the EMA has no authority to permit marketing in the different EU countries, the European Commission is the authorizing body for all centrally authorized products, who takes a legally binding decision based on EMA’s recommendation. Once granted by the European Commission, the centralized marketing authorization is valid in all EU Member States as well as in the European Economic Area countries Iceland, Liechtenstein and Norway. European Commission decisions are published in the Community Register of medicinal products for human use. Once a medicine has been authorized for use in the EU, the EMA and the EU Member States constantly monitor its safety and take action if new information indicates that the medicine is no longer as safe and effective as previously thought. The safety monitoring of medicines involves a number of routine activities ranging from: assessing the way risks associated with a medicine will be managed and monitored once it is authorized; continuously monitoring suspected side effects reported by patients and healthcare professionals, identified in new clinical studies or reported in scientific publications; regularly assessing reports submitted by the Company holding the marketing authorization on the benefit-risk balance of a medicine in real life; and assessing the design and results of post-authorization safety studies which were required at the time of authorization. The EMA can also carry out a review of a medicine or a class of medicines upon request of a Member State or the European Commission. These are called EU referral procedures; they are usually triggered by concerns in relation
to a medicine’s safety, the effectiveness of risk minimization measures or the benefit-risk balance of the medicine. The EMA has a dedicated committee responsible for assessing and monitoring the safety of medicines, the Pharmacovigilance Risk Assessment Committee. This ensures that EMA and the EU Member States can move very quickly once an issue is detected and take any necessary action, such as amending the information available to patients and healthcare professionals, restricting use or suspending a medicine, in a timely manner in order to protect patients.
Besides the Centralized procedure, pharmaceutical companies may also submit marketing authorization applications through the Decentralized procedure with the competent authority of a Member State. As the Centralized procedure is compulsory for medicines intended to treat specified diseases e.g. cancer, AIDS, neurodegenerative diseases and diabetes and optional for medicines comprising of new active substances not previously approved for the EU/EEA, in all other circumstances the Decentralized procedure should be used instead if a marketing authorization is to be obtained for several EU/EEA Member States. When following the Decentralized procedure, the applicant requests one country to be the Reference Member State (“RMS”) in the procedure. After having shared draft assessment reports to which both the applicant and the competent authorities of other Member States may respond, the Decentralized procedure results in coordinated national marketing authorization in all involved Member States. In the Mutual recognition procedure other Member States generally adopt the RMS’s assessment, unless there are important objections on the grounds of a potentially serious risk to public health. In such situations, further discussions will also be held in the Co-ordination group for Mutual recognition and Decentralized procedures (“CMDh”). When all Member States involved decide on a positive opinion on products in the CMDh, Dutch translations of the summary of product characteristics, package leaflet, labelling texts and mock-ups are submitted and a national marketing authorization is issued.
United Kingdom
In the UK, there are two main “layers” of regulation with which products containing controlled substances must comply. These are: (i) controlled drugs legislation, which applies to all products irrespective of the type of product, and (ii) the regulatory frameworks applicable to a specific category of products, in this case, pharmaceuticals and food/food supplements.
The main UK controlled drugs legislation is the Misuse of Drugs Act 1971 (the “MDA”) and the Misuse of Drugs Regulations 2001 (the “MDR”), each as amended. The MDA sets out the penalties for unlawful production, possession and supply of controlled drugs based on three classes of risk (A, B and C). The MDR sets out the permitted uses of controlled drugs based on which Schedule (1 to 5) they fall within.
In the United Kingdom, HLP003 and HLP004 are controlled as a Class A drug under the MDA and Schedule 1 drug under the MDR.
In the United Kingdom, Class A drugs are deemed to be the most dangerous, and so carry the harshest punishments for unlawful manufacture, production, possession and supply. Schedule 1 drugs can only be lawfully manufactured, produced, possessed and supplied under a controlled drugs domestic licence, which specifies the specific activities to which it relates together with any applicable conditions, issued by the UK Home Office. While exemptions do exist, none are applicable to the API. DMT is also considered a Class A drug under the MDA and as a Schedule I drug under the MDR.
The Company previously mentioned that it intended to file a clinical trial application with the UK Medical and Healthcare Products Regulatory Agency (“MHRA”) related to the HLP003 Program upon completion of its pre-clinical studies and CMC development. The Company had then decided that it
would first proceed in the U.S. and would subsequently file a clinical trial application with the MHRA. On July 17, 2025, the Company announced that it has received approval from the MHRA to commence EMBRACE, the second pivotal study in PARADIGM. Anticipated timelines related to regulatory filings are based on reasonable assumptions informed by current knowledge and information available to the Company.
Licensing Requirements
The Company obtains HLP003 API from a pharmaceutical ingredient provider who is FDA-registered and based in the United States. The API itself has been manufactured and packaged in FDA-registered facilities in the United States. The API is expected to be sent directly to the Company’s partners for research and development purposes in the United States, Canada and the UK and to its clinical trial sites in the U.S., Europe, the UK and Australia. As a part of the Asset Acquisition, the Company also acquired API. The HLP004-E API was manufactured in the Netherlands by a pharmaceutical ingredient provider that is US FDA-inspected.29
As mentioned above, in order to produce, possess and supply the API at a UK-based facility, the facility must also hold a domestic license issued by the Home Office covering the manufacture, production, possession and supply of a controlled substance. For export of the API to the United States, an export license is required for each API shipment. The export application must include details of the importer and any import license required by the local authorities in the United States. Moreover, as set out below in more detail under the heading “Pharmaceutical Products”, depending on how the API is developed and supplied, certain authorizations and licenses from the MHRA may be required to authorize some of the activities carried on at the UK-based facilities in relation to the API.
All premises that are Home Office licensed, or are intending to be licensed, in connection with the possession, and/or supply and/or production of controlled drugs should consider certain security measures.30
Typically, when controlled drugs are being transported between licensees, responsibility for their security remains with the owner and does not transfer to either the courier or the customer until the drugs arrive at their destination and are signed for. However, where a third party is involved in the transit and/or storage of controlled drugs, even if they are not the legal owners, this party also carries responsibility for their security by virtue of being ‘in possession’ of them. The MDA requires that every UK entity in possession of controlled drugs holds an appropriate Home Office License and under the Home Office guidance, each UK organization involved in the movement of controlled drugs should have a standard operating procedure covering their responsibilities, record keeping, reconciliation and reporting of thefts/losses.31
Cybin IRL Limited, the sponsor of clinical trials in the UK, is not required to hold a Controlled Drugs Licence as an Irish entity.
29 As a result of the Asset Acquisition, including the existing API, the Company did not direct the manufacturing of the API for HLP004-E and proceeded in reliance upon the representations of Entheon and the Company’s acquisition diligence. While the Company believes the HLP004-E API meets all required specifications, the Company did not oversee or direct the manufacture of the DMT API being used in HLP004-E.
30 Home Office guidance; Security guidance for all existing or prospective Home Office Controlled Drug Licensees and/or Precursor Chemical Licensees or Registrants; 2022; https://assets.publishing.service.gov.uk/media/63a1b6c8e90e075874d91825/Security_Guidance_for_all_Businesses_and_Other_Organisations_v1.5_Nov_2022.pdf
31 Home Office guidance; Guidelines for Standard Operating Procedures (SOPs); https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/480572/StandardOpProcedure.pdf.
Pharmaceutical Products
A product is regulated as a “medicinal product” under UK legislation (the Human Medicines Regulations 2012, as amended) if (i) it is a substance or combination of substances presented as having properties of preventing or treating disease in human beings (e.g., in marketing claims) or (ii) it is a substance or combination of substances that may be used by or administered to human beings with a view to (a) restoring, correcting or modifying a physiological function by exerting a pharmacological, immunological or metabolic action, or (b) making a medical diagnosis.
In respect of HLP003 and HLP004, whether a specific product restores, corrects or modifies a physiological function by exerting a pharmacological, immunological or metabolic action will depend on factors such as the concentrations of HLP003 or HLP004 (as applicable) and the mode of action of any HLP003 or HLP004 (as applicable) absorbed in the body. This requires scientific analysis.
If a product is a medicinal product, the Human Medicines Regulations 2012 require that a marketing authorization for the product granted by the UK Licensing Authority should be in place before the product is placed on the market in the UK (other, more limited, licensing options are available, such as a conditional marketing authorization, unless the product falls within one of the specified exemptions, such as supply in response to an unsolicited request from a healthcare professional to meet the special clinical needs of a particular patient under his/her care). Following the UK’s exit from the EU and the end of the transitional period, up to (and including) December 31, 2024, there were separate licensing routes and licences for products supplied: (i) in Great Britain only; (ii) in Northern Ireland only; and (iii) across the UK. From January 1, 2025, the UK Licensing Authority now licenses products across the whole of the UK through UK-wide licenses, removing the separate licensing routes for Great Britain and Northern Ireland for many medicinal products. The process for obtaining a standard marketing authorization generally involves submitting preclinical and clinical data as well as quality and manufacturing information in the form of a common technical document to the MHRA. In addition to a marketing authorization for the product itself, companies carrying out activities involving medicinal products, such as manufacturing, distribution and wholesaling, need to meet defined standards (GMP) and/or Good Distribution Practice (“GDP”) and to hold a related licence from the MHRA.
How the API is subsequently processed will determine the licences (in addition to any applicable Home Office licenses as referred to above) that the UK-based facility must hold. In particular:
•if the API is just one ‘ingredient’ (i.e. active substance) of the investigational medicinal product (the “IMP”) which is used in the clinical trial then the UK-based facility must apply to be registered with the MHRA and provide the MHRA with 60 days’ notice of the intended start of manufacture, import or distribution of the API, and comply with GMP and GDP for active substances; furthermore, an MHRA inspection may be required; and
•conversely, if the API will itself constitute the IMP, the manufacturer must, except in certain limited circumstances, hold a Manufacturer’s Authorizations for IMPs licence (“MIA(IMP)”) granted by the MHRA. In this scenario, assuming the IMP is manufactured or assembled in the UK, an MIA(IMP) would be required regardless of whether the IMP is for use in clinical trials in the UK, an EEA Member State or a third country (such as the United States or Canada). GMP, GDP and inspection requirements will apply.
Some products fall on the borderline between medicines and other categories of regulated products such as medical devices, cosmetics or food supplements. The regulatory status of the product will be determined by i) the actual effect of the product on the body; and ii) any claims made about the effect of the product. Where a product is potentially both a medicinal product and another category of product, the legal position in the UK and EU is that it will be regulated as a medicinal product.
Australia
The Australian regulatory framework for therapeutic goods (i.e. medicines) and clinical trials is overseen by the Therapeutic Goods Administration (the “TGA”).
In most cases, the Therapeutic Goods Act 1989 (Cth) (the “TG Act”) requires that medicines must be entered on the Australian Register of Therapeutic Goods before they can be imported into and/or supplied in Australia. However, in some circumstances the TGA permits access to unapproved therapeutic goods as part of clinical trials, or via a prescription from an authorized clinician.
In Australia, the regulations applicable to certain drugs are determined by their classification under the Therapeutic Goods (Poisons Standard—October 2025) Instrument 2025 (Poisons Standard), a legislative instrument made under the TG Act and maintained by the TGA.
The Poisons Standard is given legal effect and enforced through State and Territory legislation, regulations and instruments across Australia, including:
•Australian Capital Territory: Medicines, Poisons and Therapeutic Goods Act 2008 (ACT); Medicines, Poisons and Therapeutic Goods Regulation 2008 (ACT);
•New South Wales: Poisons and Therapeutic Goods Act 1966 (NSW); Poisons and Therapeutic Goods Regulation 2008 (NSW);
•Northern Territory: Medicines, Poisons and Therapeutic Goods Act 2012 (NT); Medicines, Poisons and Therapeutic Goods Regulations 2014 (NT);
•Queensland: Medicines and Poisons Act 2019 (Qld); Medicines and Poisons (Poisons and Prohibited Substances) Regulation 2021 (Qld); Medicines and Poisons (Medicines) Regulation 2021 (Qld); Medicines and Poisons (Pest Management Activities) Regulation 2021 (Qld);
•South Australia: Controlled Substances Act 1984 (SA); Controlled Substances (Poisons) Regulations 2011 (SA); Controlled Substances (Controlled Drugs, Precursors and Plants) Regulations 2014 (SA);
•Tasmania: Poisons Act 1971 (Tas); Poisons Regulations 2018 (Tas);
•Victoria: Drugs, Poisons and Controlled Substances Act 1981 (Vic) and Drugs, Poisons and Controlled Substances Regulations 2017 (Vic); and
•Western Australia: Medicines and Poisons Act 2014 (WA); Medicines and Poisons Regulations 2016 (WA).
Schedule 8 of the Poisons Standard lists controlled drugs which are only available for therapeutic use via a prescription for certain indications. These drugs are subject to more stringent controls than other prescription medicines because of the relatively greater risk of misuse. Schedule 9 of the Poisons Standard contains prohibited substances which cannot be used without regulatory approvals (for example, approvals for use in clinical trials) because these substances have been deemed to have a higher risk of abuse, misuse or diversion.
HLP003 and HLP004 are prohibited substances under Schedule 9 of the Poisons Standard. The use and supply of these substances is only permitted under the TGA’s Authorized Prescriber Scheme, the Special Access Scheme and clinical trials approved by, or notified to, the TGA.
The importation of schedule 9 drugs is prohibited and the importation an/or supply of a prohibited substance without the relevant approvals is a criminal offence. Approvals must be obtained from the TGA and the Office of Drug Control to import HLP003 for the purpose of conducting clinical trials.
Under section 19 of the TG Act, the TGA may grant approval for the importation and/or supply of unapproved therapeutic goods to a clinician for use in the treatment of a patient, or solely for experimental purposes in humans. The Office of Drug Control also regulates the importation of psychotropic substances and a licence must be issued by the Narcotics Control Section in order to import HLP003 into Australia. Individual permits must then be obtained for each consignment of HLP003. Further licences and/or permits must be sought from the State or Territory health authority in the State or Territory in which the trial is to be conducted, in order to manufacture, possess, supply or use schedule 9 drugs for clinical trials. The specific regulations applicable will depend on the State or Territory in which these acts occur.
By way of example, in New South Wales section 17D of the Poisons and Therapeutic Goods Act 1966 (NSW) enables the Secretary of the New South Wales Ministry of Health to authorize a person to manufacture, possess, use or supply a specified Schedule 9 substance. The conditions of any authorization obtained in New South Wales can also include additional restrictions on the personnel authorized to use Schedule 9 substances in the trial and further storage, handling and disposal requirements. Meanwhile, in the state of Victoria, an additional licence/permit is also required to import schedule 9 drugs.
Clinical trials
All clinical trials conducted in Australia must have a local sponsor (an overseas company cannot be the sponsor of a clinical trial conducted in Australia). The trial sponsor is responsible for the initiation, management and financing of the trial and carries the legal risk arising from the conduct of the trial. The sponsor must also ensure that the use of therapeutic goods in the trial must be in accordance with the Guideline for Good Clinical Practice, the National Statement on Ethical Conduct in Research Involving Humans and the protocol approved by the HREC responsible for monitoring the conduct of the trial.
Clinical trials involving unapproved therapeutic goods, including products containing HLP003, may be conducted in Australia under two schemes: Clinical Trial Notification (the “CTN”) or Clinical Trial Approval (the “CTA”). The Australian clinical trial involving HLP003 was approved through the CTN scheme, obtained clearance from multiple Ethics Committees of the TGA, and the study site Research Governance Offices.
Under the CTN scheme the TGA does not review or evaluate any data relating to clinical trials. All material relating to the proposed trial, including the trial protocol is submitted directly to, and reviewed by the HREC and the site where the trial will be conducted. The CTN scheme can be used where a foreign regulator with comparable regulatory requirements has already approved a clinical trial for an equivalent indication.
A clinical trial is deemed to be notified by the TGA, after the CTN form has been submitted to the TGA and the relevant fee has been paid. Once that occurs, the CTN exemption comes into effect and the trial sponsor can lawfully supply the goods.
Under the CTA scheme, the TGA will review and evaluate relevant, but limited, scientific data (which may be preclinical and early clinical data) prior to the start of a trial. The TGA’s primary responsibility is to review the safety of the product and the HREC is responsible for considering the scientific and ethical issues of the proposed trial protocol. Conduct of a clinical trial under the CTA scheme must also be approved by the responsible HREC.
Throughout the conduct of the trial, sponsors must ensure that any advertisements and promotional materials relating to the trial do not promote the use of unapproved therapeutic goods. However, materials promoting clinical trials directly to clinicians fall outside of these restrictions.
Upon the completion of the Australian trial, it will not be possible to make an application to register HLP003 on the Australian Register of Therapeutic Goods because non-deuterated HLP003 is currently listed in schedule 9 of the Poisons Standard. However, this may be possible if an application is made to the TGA to change the scheduling of HLP003 so that it is removed from Schedule 9 of the Poisons Standard.
In some circumstances it is possible for clinicians to import and then supply unapproved therapeutic goods, including schedule 9 drugs, to patients. The Special Access Scheme (the “SAS”) permits approved clinicians to prescribe Schedule 9 drugs to individual patients in limited circumstances. Category A of the SAS enables medical practitioners to access unapproved therapeutic goods for a patient who is seriously ill. Clinicians who are unable to access HLP003 through the Category A can also seek approval to access HLP003 for patients on a case by case basis under Category B of the SAS.
Meanwhile, medical practitioners who wish to prescribe HLP003 to more than one patient can seek to become an authorized prescriber. The medical practitioner must first obtain approval for the proposed use of HLP003 from a human research ethics committee (because the TGA does not consider that non-deuterated forms of HLP003 have an established history of use) and then make an application to the TGA.
Poland
The manufacture, trade, processing, possession, use and distribution of narcotic drugs, psychotropic substances and new psychoactive substances in Poland are governed primarily by two legal acts: the Act of July 29, 2005 on Counteracting Drug Addiction (the “ACDA”), and the Act of September 6, 2001 the Pharmaceutical Law (the “PLA”).
Under the ACDA, psychotropic substances are defined as substances of natural or synthetic origin, in pure form or in the form of a preparation, acting on the central nervous system.
Psychotropic substances are divided into four groups: I-P, II-P, III-P and IV-P. Narcotic drugs are analogously divided into groups I-N to IV-N. The classification is based on the risk of abuse and dependence and the extent of recognized medical use.
According to the ACDA narcotic drugs of groups I-N and II-N and psychotropic substances of groups II-P, III-P and IV-P may only be used for medical, industrial or research purposes. Psychotropic substances
of group I-P may be used exclusively for scientific research purposes. This is due to the fact that psychotropic substances classified to group I-P substances are considered the most dangerous, with a high potential for abuse and no accepted medical use under current Polish law.
HLP003 and HLP004 are strictly controlled substances. According to the Ordinance of the Minister of Health of 17 August 2018 on the list of psychotropic substances, narcotic drugs and new psychoactive substances (Rozporządzenie Ministra Zdrowia z dnia 17 sierpnia 2018 r. w sprawie wykazu substancji psychotropowych, środków odurzających oraz nowych substancji psychoaktywnych), HLP003 and HLP004 are classified as group I-P psychotropic substances. Thus, for any product containing HLP003 or any group I-P psychotropic substances to be available for commercial marketing in Poland, such substance must be first reclassified/reassigned to another group.
Only strictly defined entities specified in the ACDA may possess narcotic drugs, psychotropic substances and new psychoactive substances or their preparations, i.e. entrepreneurs holding appropriate permits, scientific institutions, or entities additionally specified in Regulation 273/2004 or Regulation 111/2005. Undertaking activities involving the manufacture, processing, conversion, import, or distribution of narcotic drugs or psychotropic substances requires an appropriate authorization issued by the national authorities. Authorization is also required for the manufacture, processing, or conversion of such substances for the purpose of conducting scientific research by scientific institutions within the scope of their statutory activities, in relation to narcotic drugs of groups I-N, II-N, and IV-N, or psychotropic substances of groups I-P, II-P, III-P, and IV-P. A separate authorization is additionally required for the use of narcotic drugs or psychotropic substances for the purpose of conducting scientific research by scientific institutions within the scope of their statutory activities.
Obtaining the required authorizations involves meeting numerous criteria set out in detail in the ACDA and in the Ordinance of the Minister of Health of 9 November 2015 on the issuance of permits for the manufacture, processing, conversion, import, distribution or use for scientific research purposes of narcotic drugs, psychotropic substances or category 1 precursors (Rozporządzenie Ministra Zdrowia z dnia 9 listopada 2015 r. w sprawie wydawania zezwoleń na wytwarzanie, przetwarzanie, przerabianie, przywóz, dystrybucję albo stosowanie w celu prowadzenia badań naukowych środków odurzających, substancji psychotropowych lub prekursorów kategorii 1).
The criteria include, among others: maintaining adequate procedures and internal control systems, employing a qualified person responsible for supervision of narcotics and psychotropics, ensuring physical security of premises (two-lock doors, alarm system, reinforced windows, safes or fixed metal cabinets), maintaining inventory and turnover records. In the case of conducting scientific research, it is also necessary to:
•define the purpose and scope of the research, as well as the methods for sample preparation and analysis of research results;
•submit to the regulatory authority a copy of the institution’s statute and a commitment to provide copies of any amendments to the statute;
•specify the planned start and end dates of the research; and
•in the case of manufacture, processing, or conversion of narcotic drugs of groups I-N, II-N, and IV-N or psychotropic substances of groups I-P, II-P, III-P, and IV-P, maintain documentation of technically justified standards for the consumption of raw materials used in the process, as well as the acceptable loss rates at each stage of production.
The records of narcotic drugs, psychotropic substances, and precursors must be kept in a way that allows easy tracking of all events and transactions related to them, especially those involving the receipt and release of these substances. A separate control book must be kept for each narcotic and psychotropic substance (by group and dosage), recording receipts, issues, stock levels, and remarks. Entries are made by qualified person. The control book is retained for five years from the beginning of the year following the last entry.
Unlawful manufacturing, processing or conversion of narcotic drugs or psychotropic substances is subject to imprisonment for up to three years. In the case of significant quantities of narcotic drugs or psychotropic substances, the perpetrator is liable to a fine and imprisonment for three to 20 years. Narcotic drugs, psychotropic substances, new psychoactive substances, or their preparations, as well as category 1 precursors, possessed without authorization, are subject to seizure by law enforcement or customs authorities in the manner specified in criminal procedure. Sized products are subject to forfeiture of the goods to the benefit of the State Treasury.
Clinical (scientific) research involving psychotropic substances is regulated by several legal acts, including the ACDA, the PLA, the Act of March 9, 2023, on clinical trials of medicinal products used in humans, Regulation (EU) No 536/2014 of April 16, 2014, on clinical trials on medicinal products for human use and repealing Directive 2001/20/EC, as well as implementing acts and regulations, including the Ordinance of the Minister of Health of November 9, 2015, on the issuance of permits for the manufacture, processing, conversion, import, distribution, or use for scientific research purposes of narcotic drugs, psychotropic substances, or category 1 precursors. Only entities authorized for this purpose may conduct scientific studies involving psychotropic substances from group I-P, including, for example, universities, scientific institutes and research institutes.
Clinical trials involving psychotropic substance are legally possible only as scientific research, and only after obtaining all of the following approvals, including authorization to use the I-P psychotropic substance for research, positive opinion of a Bioethics Committee, clinical-trial authorization, as well as all permits required for handling a controlled substance, such as authorizations for manufacture, processing, conversion, import, export, intra-EU acquisition, and intra-EU supply of narcotic drugs and psychotropic substances.
All stages of the study must comply with the Good Clinical Practice (“GCP”) standards, relevant EU and national data protection, pharmacovigilance, and biosafety regulations, and the ethical principles set out in the Declaration of Helsinki. Given the high risk of abuse of group I-P substances, the scientific institution must maintain secure storage conditions (in line with the standards included in the Ordinance of the Minister of Health of November 9, 2015), keep detailed logs of the quantity, source, and use of each psychotropic substance, and report any discrepancies or losses immediately to the competent authorities.
Under the current ACDA, medicinal products containing group I-P substances cannot be granted marketing authorization, as the use of such substances is permitted only for research purposes. This prohibition effectively prevents them from being included in authorized medicinal products. Obtaining a marketing authorization would thus require first a reclassification of the substance (from group I-P to II-P or lower). Medicinal products containing psychotropic substances may be authorized and marketed in Poland only if they meet the definition of a medicinal product under the PLA and comply with all ACDA restrictions applicable to controlled substances. According to the PLA, a medicinal product is any substance or combination of substances presented for preventing or treating disease in humans or animals, or administered with a view to making a diagnosis or restoring, correcting or modifying physiological functions by exerting a pharmacological, immunological or metabolic action.
Marketing authorization of a medicinal product can be obtained through several procedures, depending on the product type and the geographical scope:
•National Procedure – authorization is granted by the national authority - the Office for Registration of Medicinal Products. The authorization is valid only in Poland.
•Decentralised Procedure – used for products not yet authorized in any EU Member State. The applicant applies simultaneously in several countries, with one acting as the Reference Member State (“RMS”).
•Mutual Recognition Procedure – used when a product is already authorized in one EU Member State. Other countries recognize this authorization based on the RMS assessment.
•Centralised Procedure – conducted by the EMA. Once approved by the European Commission, the authorization is valid across all EU and EFTA countries.
Medicinal products that have obtained a marketing authorization issued by the President of the Office for Registration of Medicinal Products, Medical Devices and Biocidal Products may be lawfully placed on the market.
The application for a marketing authorization of a medicinal product must include documentation confirming its manufacturing process, quality control, safety, and efficacy. Specifically:
•description of manufacturing process and quality control methods,
•written confirmation of GMP audit for the active substance manufacturer,
•information on storage, dispensing, and disposal, including environmental risk assessment,
•results and summaries of pharmaceutical, non-clinical, and clinical studies,
•summary of the pharmacovigilance system, including declarations from the marketing authorization holder and details of the qualified person,
•risk management plan for the product’s use,
•ethical compliance statement for clinical trials conducted outside the EU/EFTA,
•expert declarations of qualifications,
•summary of Product Characteristics,
•mock-ups of packaging and patient leaflet, with a readability test report,
•copies of authorizations, decisions, and related documents from EU, EFTA, or third countries,
•list of countries where the application is under review, and
•copy of the manufacturing authorization from the production site.
By way of a separate national regulation, the Minister of Health indicates psychoactive substances and their maximum content in medicinal products necessary for effective treatment within the acceptable period of safe treatment for one person. This obligation is intended to limit the dispensing of medicinal products in single sales, with a view to protecting public health and the safety and efficacy of medicinal products, as well as their dosage.
Greece
The Company’s development activities in Greece relate to its multinational pivotal clinical program evaluating HLP003 for the potential adjunctive treatment of MDD (EMBRACE). The Company has received European approval to initiate the EMBRACE study in selected EU Member States, including
Greece, and will conduct any activities in Greece in accordance with applicable EU and Greek legal and regulatory frameworks governing investigational medicinal products and controlled substances.
In Greece, controlled substances are regulated under national legislation implementing the UN Single Convention on Narcotic Drugs (1961), the UN Convention on Psychotropic Substances (1971) and related EU law, including Greek Law 4139/2013 on addictive substances and Presidential Decree 148/2007 (codification of provisions of national drugs legislation). HLP003, as a controlled substance, is subject to strict prohibitions on manufacture, possession, distribution, import and export, unless expressly authorized for scientific or very limited medical purposes by duly authorized persons or entities, operating under licence and subject to stringent security, recordkeeping and oversight requirements. Licensing and enforcement responsibilities are shared between the Ministry of Health (Department for Narcotic and Psychotropic Substances) and the Greek National Organization for Medicines (the “EOF”). As an EU Member State, Greece applies Regulation (EU) No. 536/2014 on clinical trials of medicinal products for human use (the Clinical Trials Regulation; “CTR”), including the Clinical Trials Information System for submissions, supervision and transparency. The EOF and the competent National Ethics Committee are responsible, within their respective remits, for authorization and ethical review of clinical trials conducted in Greece.
Clinical trials in Greece involving controlled substances require all standard clinical trial approvals under the EU Clinical Trials Regulation, together with any specific national licences and permits applicable to the handling, storage, receipt, use, transport, import and export of the relevant controlled substances at trial sites and other facilities involved in the conduct of the study. Compliance includes, among other obligations, appropriate physical security, access controls, inventory reconciliation, documentation and retention, diversion prevention and incident reporting. Additionally, import and export of controlled substances are subject to prior authorization and documentation by the competent authorities pursuant to Law 4139/2013 and the relevant codified provisions under Presidential Decree 148/2007, as well as the requirements of the exporting and importing jurisdictions.
Helus Pharma does not itself manufacture, handle or distribute controlled substances in Greece. The Company sponsors and oversees clinical research conducted by licensed third-party institutions and clinical sites, that are responsible for obtaining and maintaining all applicable Greek licences, permits and authorizations for controlled substances and investigational medicinal products, and for ensuring site-level compliance with the Clinical Trials Regulation, current GCP standards, and Greek controlled drug requirements under Law 4139/2013, including security, storage, documentation, and reporting obligations. Helus Pharma continues to monitor compliance through its quality and clinical governance systems and relies on contractual undertakings, ongoing oversight, and representations and warranties from its partners and vendors with respect to regulatory compliance.
Any future commercialization of the HLP003 product would require marketing authorization (“MA”) and continued compliance with applicable controlled-substance controls, pharmacovigilance, labelling, GDP/GMP and other requirements. Pharmaceutical products in Greece are regulated primarily under the EU and national frameworks implementing Directive 2001/83/EC and Regulation No. 726/2004. Products authorized through the EU centralized procedure receive a single marketing authorization issued by the European Commission, which is automatically valid in all EU Member States, including Greece.
While the centralized MA permits marketing throughout the EU, national-level compliance obligations remain in place, including:
•Pricing and reimbursement procedures overseen by the Ministry of Health and EOF;
•Notification to EOF prior to product launch in the national market;
•Pharmacovigilance and safety reporting obligations in accordance with Regulation (EU) No. 1235/2010 and Directive 2010/84/EU;
•GMP and GDP requirements, implemented through Greek secondary legislation (Ministerial Decision ΔΥΓ3α/Γ.Π. 32221/2013);
•Greek-language labelling and patient information leaflet requirements; and
•Restrictions on advertising and promotion, as applicable.
For centrally authorized medicinal products containing controlled substances, additional Greek licensing and notification requirements apply for importation, distribution, and storage, consistent with the national controlled substance framework under Law 4139/2013. EOF may require confirmation of appropriate site licences prior to allowing local supply chain activities. To be noted that changes to Greek or EU legislation, regulations, scheduling or enforcement priorities could increase compliance burdens, delay clinical timelines, or adversely affect the Company’s operations in Greece.
As of the date of this MD&A, the Company is in compliance with applicable EU and Greek laws and the related licensing frameworks, as they pertain to the Company’s activities in Greece conducted through authorized third parties. The Company and, to its knowledge, its third party researchers and suppliers have not received any notices of violation that may have a material impact on the Company’s licences, business activities or operations in Greece.
Ireland
In Ireland, HLP003 is a controlled substance under the Misuse of Drugs Act, 1977, 1984 and 2015 (the “Ireland MDA”), the Misuse of Drugs Regulations 2017 (the “Ireland MDR”) and the Criminal Justice (Psychoactive Substances) Act 2010 (the “2010 Act”). These are the primary legislative instruments which govern controlled substances in Ireland. This legislation regulates the use, possession, supply, licensing, and administration of listed scheduled substances and establishes the offences and penalties for infringement of the legislation.
Any substance, product or preparation (whether natural or otherwise) containing HLP003 is classed as a Schedule 1 controlled substance under the Ireland MDA and the Ireland MDR. Accordingly, HLP003 is subject to the strict regime of control that applies.
As a Schedule 1 controlled substance under the Ireland MDA, unlawful manufacturing, production, preparation, importation, exportation, supply, or distribution of Schedule 1 controlled substances carries onerous obligations and harsh punishments for contravention; this includes prohibition orders, closure orders, fines and/or terms of imprisonment of up to 14 years. The Gardaí and Customs officials are granted powers to search persons, vehicles, premises and postal packages suspected of possessing/containing a Schedule 1 controlled substance and/or a psychoactive substance for human consumption.
Pursuant to section 14 of the Ireland MDA, in certain circumstances, the Minister for Health “may grant licences or issue permits or authorizations for any of the purposes of this Act, attach conditions to any such licence, permit or authorization, vary such conditions and revoke any such licence, permit or authorization”. Where licences are granted, there are very strict conditions imposed on licence holders. For example, strict conditions can be placed regarding the security, storage and documenting controlled substances. Further, the 2010 Act permits the Minister to make an order declaring that the Act shall not
apply in relation to any “substance, product, preparation, plant, fungus or natural organism” as specified in the order.
The Irish Government’s Legislative Programme for Autumn 2025 does not contain any proposed amendments to the above-mentioned legislation which currently govern controlled substances.
The Company has received European Clinical Trial Application approval to initiate the EMBRACE study in Ireland and, as such, will have to abide by the regulations governing clinical trials in Ireland. EU Regulation 536/2014 (the “CTR”) is implemented in Ireland via the EU (Clinical Trials on Medicinal Products for Human Use) (Principal) Regulations 2022 (S.I. No. 99 of 2022), the EU (Clinical Trials on Medicinal Products for Human Use) (National Research Ethics Committees) Regulations 2022 (S.I. No. 41 of 2022), and the EU (Clinical Trials on Medicinal Products for Human Use) Regulations 2022 (S.I. No. 40 of 2022), (together, the “2022 Regulations”).
Clinical trials taking place in Ireland must be conducted in accordance with the CTR, the 2022 Regulations, the Declaration of Helsinki, clinical trial authorization (which has been granted), CT protocol(s), any accompanying documentation relevant to the clinical trial and the conditions and principles of good clinical practice. The manufacturing and importation of an investigational medicinal product in Ireland requires a manufacturer’s authorization.
Schedule 1 controlled substances can be used for “research, forensic analysis or use as an essential intermediate or starting material in an industrial manufacturing process” once a Ministerial licence under section 14 of the MDA has been obtained. Where the controlled substance is so licensed and the proposed trial is fully compliant with the CTR and the 2022 Regulations, it is possible for a Schedule 1 controlled substance to be used in clinical trials in Ireland.
The clinical trial sponsor (i.e., the Company) must also upload the clinical trial protocol, the investigator’s brochure, the assessment report and any inspection reports to the EU-wide Clinical Trials Information System (“CTIS”). Personal and commercially sensitive/confidential information may be redacted from these documents prior to upload as once they are posted to the CTIS they will be publicly available on the CTIS website.
Canada
In Canada, oversight of healthcare is divided between the federal and provincial governments. The federal government is responsible for regulating, among other things, the approval, import, sale, and marketing of drugs, whether natural or novel. The provincial/territorial level of government has authority over the delivery of health care services, including regulating health facilities, administering health insurance plans such as the Ontario Health Insurance Plan, distributing prescription drugs within the province, and regulating health professionals such as doctors, psychologists, psychotherapists and nurse practitioners. Regulation is generally overseen by various colleges formed for that purpose, such as the College of Physicians and Surgeons of Ontario.
Certain compounds, such as HLP003, are considered controlled substances under Schedule III of the CDSA. In order to conduct any scientific research, including preclinical and clinical trials, using compounds listed as controlled substances under Schedule III of the CDSA, an exemption under Section 56 of the CDSA (“Section 56 Exemption”) is required.
Health Canada has not approved HLP003 as a drug for any indication. However, there are legal routes through which HLP003 may be accessed for medical or scientific purposes. The Canadian Minister of
Health can grant exemptions under section 56 of the CDSA to use controlled substances if it is deemed to be necessary for a medical or scientific purpose or is otherwise in the public interest. The Company has not applied for a Section 56 Exemption from Health Canada. Health Canada’s Special Access Program (“SAP”) was designed to provide Canadians to access certain restricted drugs before they are formally approved for use in Canada. In January 2022, certain amendments to the SAP came into force to permit medical practitioners treating patients with serious or life-threatening conditions to request access to restricted drugs that have not yet been approved for sale in Canada when conventional therapies have failed, are unsuitable, or unavailable in Canada. Such amendments create a means of legally accessing HLP003 through the SAP. The Company has not applied for access under the SAP.
The possession, sale or distribution of controlled substances is prohibited unless specifically permitted by the government. A party may seek government approval for a Section 56 Exemption to allow for the possession, transport or production of a controlled substance for medical or scientific purposes. Products that contain a controlled substance such as HLP003 cannot be made, transported or sold without proper authorization from the government. A party can apply for a Dealer’s Licence under the Food and Drug Regulations (Part J). In order to qualify as a licensed dealer, a party must meet all regulatory requirements mandated by the regulations including having compliant facilities, compliant materials and staff that meet the qualifications under the regulations of a senior person in charge and a qualified person in charge. Assuming compliance with all relevant laws (Controlled Drugs and Substances Act, Food and Drugs Regulations) and subject to any restrictions placed on the licence by Health Canada, an entity with a Dealer’s Licence may produce, assemble, sell, provide, transport, send, deliver, import or export a restricted drug (as listed in Part J in the Food and Drugs Regulations – which includes HLP003) (see s. J.01.009 (1) of the Food and Drug Regulations).
The Company intends to sponsor and work with licensed third parties to conduct any clinical trials and research and does not handle controlled substances. If the Company were to conduct this work without the reliance on third parties, it would need to obtain additional licences and approvals described above.
Research and Development
The process required before a prescription drug product candidate may be marketed in Canada generally involves:
•Chemical and Biological Research - Laboratory tests are carried out on tissue cultures and with a variety of small animals to determine the effects of the drug. If the results are promising, the manufacturer will proceed to the next step of development.
•Pre-clinical Development – Animals are given the drug in varying amounts over differing periods of time. If it can be shown that the drug causes no serious or unexpected harm at the doses required to have an effect, the manufacturer will proceed to clinical trials.
•Clinical Trials — Phase 1 - The first administration in humans is to test if people can tolerate the drug. If this testing is to take place in Canada, the manufacturer must prepare a clinical trial application for the Therapeutic Products Directorate of Health Canada (the “TPD”). This includes the results of the first two steps and a proposal for testing in humans. If the information is sufficient, the Health Products and Food Branch of Health Canada (the “HPFB”) grants permission to start testing the drug, generally first on healthy volunteers.
•Clinical Trials — Phase 2 - Phase 2 trials are carried out on people with the target condition, who are usually otherwise healthy, with no other medical condition. Trials carried out in Canada must
be approved by the TPD. In phase 2, the objective of the trials is to continue to gather information on the safety of the drug and begin to determine its effectiveness.
•Clinical Trials — Phase 3 - If the results from phase 2 show promise, the manufacturer provides an updated clinical trial application to the TPD for Phase 3 trials. The objectives of phase 3 include determining whether the drug can be shown to be effective, and have an acceptable side effect profile, in people who better represent the general population. Further information will also be obtained on how the drug should be used, the optimal dosage regimen and the possible side effects.
•New Drug Submission - If the results from phase 3 continue to be favourable, the drug manufacturer can submit a new drug submission (“NDS”) to the TPD. A drug manufacturer can submit an NDS regardless of whether the clinical trials were carried out in Canada. The TPD reviews all the information gathered during the development of the drug and assesses the risks and benefits of the drug. If it is judged that, for a specific patient population and specific conditions of use, the benefits of the drug outweigh the known risks, the HPFB will approve the drug by issuing a notice of compliance.
Compliance with Applicable Laws
The Company oversees and monitors compliance with applicable laws in each jurisdiction in which it operates. In addition to the Company’s senior executives and the employees responsible for overseeing compliance, the Company has local counsel engaged in every jurisdiction in which it operates and has received legal opinions or advice in each of these jurisdictions regarding (a) compliance with applicable regulatory frameworks, and (b) potential exposure to, and implications arising from, applicable laws in jurisdictions in which the Company has operations or intends to operate.
The Company works with third parties who require regulatory licensing to handle scheduled drugs. The Company continuously updates its compliance and channel programs to maintain regulatory standards set for drug development. The Company also works with clinical research organizations who maintain batch records and data storage for the Company’s clinical programs.
Additionally, the Company has established a Medical & Clinical Advisory Team, a Research, Clinical and Regulatory Team and a Government Relations and Communications Team with cross-functional expertise in business, neuroscience, pharmaceuticals, mental health and serotonergic agonists to advise management.
In conjunction with the Company’s human resources and operations departments, the Company oversees and implements training on the Company’s protocols. The Company will continue to work closely with external counsel and other compliance experts, and is evaluating the engagement of one or more independent third party providers to further develop, enhance and improve its compliance and risk management and mitigation processes and procedures in furtherance of continued compliance with the laws of the jurisdictions in which the Company operates.
The programs currently in place include monitoring by executives of the Company to ensure that operations conform to and comply with required laws, regulations and operating procedures. The Company is currently in compliance with the laws and regulations in all jurisdictions and the related licensing framework applicable to its business activities.
The Company and, to its knowledge, each of its third-party researchers, suppliers and manufacturers have not received any non-compliance, citations or notices of violation which may have an impact on the Company’s licences, business activities or operations.
The Company conducts due diligence on third-party researchers, medical professionals, clinics, cultivators, processors and others as applicable, with whom it engages. Such due diligence includes but is not limited to the review of necessary licenses and the regulatory framework enacted in the jurisdiction of operation. Further, the Company generally obtains, under its contractual arrangements, representations and warranties from such third parties pertaining to compliance with applicable licensing requirements and the regulatory framework enacted in the jurisdiction of operation.
Patent Cooperation Treaty
The Patent Cooperation Treaty (“PCT”) facilitates filing for patent recognition in multiple jurisdictions simultaneously using a single uniform patent application. 158 countries, including Canada and the United States have ratified the PCT.
Ultimately, patents are still granted in each country individually. As such, the PCT procedure consists of two phases: filing of an international application, and national evaluation under the patent laws in force in each country where a patent is sought.
Within 12 months of filing a provisional patent application at the United States Patent and Trademark Office, the Company may elect to file a regular utility patent application in the United States in tandem with filing a PCT application with the World Intellectual Property Office, in each case claiming priority to the provisional patent application. Within 30 months of the provisional filing date, deadlines begin for a PCT application to enter the national phase in desired jurisdictions globally, such as Canada (30 months) and Europe (31 months), in each case claiming priority to the provisional patent application.
While the Company is focused on programs using compounds that are regulated as controlled substances in many jurisdictions, the Company does not have any direct or indirect involvement with the illegal selling, production or distribution of any substances in the jurisdictions in which it operates. The Company is exploring drug development within approved laboratory clinical trial settings conducted within approved regulatory frameworks. Though highly speculative, should any prescription drug product be developed by the Company (which, if it does occur, would not be for several years), such drug product will not be commercialized prior to receipt of applicable regulatory approval, which will only be granted if clinical evidence of safety and efficacy for the intended use(s) is successfully developed. The Company may also employ non-prescription drugs, where appropriate.
Business Objectives of the Company
Key elements of the Company’s growth strategy include: (i) progressing its NSA division through the development and commercialization of key NSA molecules (including tryptamines and phenethylamines) and delivery mechanisms; (ii) working to develop the synthetic production of HLP003 active pharmaceutical ingredients; (iii) obtaining regulatory approval for HLP003 targeting MDD; (iv) establishing strategic partnerships to advance its scientific research and to develop patented or trade secret intellectual property for the Company’s NSAs and processes related to NSAs; and (v) sponsoring clinical studies to determine the safety and efficacy of delivery mechanisms, chemically synthesized NSA compounds and screening protocols.
Production and Raw Materials
The Company has established contractual sources of synthetic GMP and non-GMP raw materials to support its development operations through licensed third-party suppliers located in Canada, the United States, the UK and Europe. Such raw materials are expected to be, in general, readily available and in adequate supply to meet the Company’s need for development quantities, or custom manufactured on the Company’s behalf.32 The prices of research quantities of novel tryptamine compounds are generally higher than commercial supply prices at significantly larger scale and the Company, therefore, expects its supply prices to reduce over time. Development and production of the Company’s proprietary novel compounds is performed under confidential contractual agreements.
Foreign Operations
The Company’s management is located in Canada, Ireland, the United Kingdom and the United States led by others in local jurisdictions. The Company’s raw materials are expected to be sourced from a supplier in the United States and are expected to be manufactured and packaged in FDA registered facilities in the United Kingdom. Such raw materials are expected to be sent directly to the Company’s partners for research and development purposes pursuant to its corresponding agreements, subject to receipt of all necessary approvals.
The Company conducts its international operations to conform to local variations, economic realities, market customs, consumer habits and regulatory environments. The Company will modify its products (including labeling of such products) and its distribution and marketing programs in response to local and foreign legal requirements and customer preferences.
The Company’s international operations are subject to many of the same risks that its domestic operations face. These include competition and the strength of the relevant economy. In addition, international operations are subject to certain risks inherent in conducting business abroad, including foreign regulatory restrictions, fluctuations in monetary exchange rates, import-export controls and the economic and political policies of foreign governments. Government regulations in foreign countries may prevent or delay the introduction, or require the reformulation, of certain of its products. Compliance with such foreign governmental regulations is generally the responsibility of the Company’s distributors in those countries. These distributors are independent contractors whom the Company does not control. The importance of these risks increases as the Company’s international operations grow and expand. See “Risk Factors”.
Market for Products
Market Segment, Market Acceptance and Geographic Areas
The Company is focused on developing novel compounds and improving the bioavailability and pharmacokinetic profiles of existing compounds to target psychiatric and neurological conditions. The Company is focused on progressing its NSA compounds, delivery mechanisms and supportive treatment platforms.
32 At this time the Company has not entered into commercial supply agreements and has no control over price or conditions. The Company has assumed that it will be able to enter into commercial supply agreements at such a time when there will be sufficient competition in the market which will render prices reasonable.
Specialized Skills and Knowledge
The Company’s directors and officers possess a wide range of professional skills and experience relevant to pursuing and executing on the Company’s business strategy. Drawing on significant experience in various industries and sectors, the Company believes its management has a demonstrated track record of bringing together all of the key components for a successful pharmaceutical company, such as strong technical skills, expertise in planning and financial controls, ability to execute on business development opportunities, and capital markets expertise. The operational skills of the Company’s management include valuable knowledge and ability to analyze demographics and consumer purchasing habits, and tailor product brands and consumer retail experiences based on relevant demographic data.
By leveraging the strengths and experiences of its management team (i.e., individuals who possess a wealth of combined knowledge and experience necessary for the research and development, sales, marketing, and distribution of pharmaceutical products) the Company intends to, over time, establish itself as a leader in the NSA pharmaceutical industry. The Company will continue to build out its team with specialists on an “as-needed” basis.
The Company’s current directors, officers and key executives have significant collective experience with NSAs, medicinal chemistry, pre-clinical and clinical operations, clinical psychology, quality and regulatory affairs, in addition to a track record of growing pharmaceutical companies including aspects of commercial operations, securities and capital markets. Collectively, the Company believes that it has adequate access to the current and future skill sets required to grow and sustain its business.
Cyclical or Seasonality of Business
The Company’s business is not expected to be cyclical or seasonal.
Employees
At the current stage of development, the Company is focused on maintaining a lean corporate structure, utilizing a highly experienced core team of senior executives and managers, while leveraging a cost-effective ecosystem of independent contractors, consultants and advisors, on an “as needed” basis. The Company employs less than 100 current full-time staff.
Intellectual Property
Helus Pharma has title to twenty eight granted US patents and ninety one granted national (non-US) patents, including claims directed to compositions of matter and methods of use in support of its research and development and preclinical and clinical trial programs. Granted European patents are counted as a single granted patent (as opposed to multiple patents in each European territory in which the patent is in force). Certain of the Company’s patent rights may be subject to out-license and option arrangements, which may limit Helus Pharma’s ability to use or further out-license such patent rights.
| | | | | |
| Patent Number | Jurisdiction of Filing |
| 11,242,318 | United States |
| 11,724,985 | United States |
| 11,746,088 | United States |
| 11,834,410 | United States |
| 11,958,807 | United States |
12,110,272 | United States |
| | | | | |
| Patent Number | Jurisdiction of Filing |
12,240,813 | United States |
12,291,499 | United States |
7766623 | Japan |
7770467 | Japan |
| 312785 | Israel |
12,122,741 | United States |
7802769 | Japan |
| 2021327136 | Australia |
| 12603504 | United States |
| 2018311307 | Australia |
| 2020378647 | Australia |
| 2020381103 | Australia |
| 2021334933 | Australia |
| 2021204158 | Australia |
| 2020286709 | Australia |
1120220221983 | Brazil |
| 3104072 | Canada |
| 3160337 | Canada |
| 3179161 | Canada |
3142290 | Canada |
| 3160334 | Canada |
| 3179335 | Canada |
| ZL202080087091.0 | China |
| ZL202080087092.5 | China |
| ZL202180044031.5 | China |
| ZL202180046463.X | China |
| ZL202080050439.9 | China |
ZL202180090269.1 | China |
| 046951 | Eurasian Patent Office |
| 048675 | Eurasian Patent Office |
| 049402 | Eurasian Patent Office |
| 049106 | Eurasian Patent Office |
| 3463323 | European Patent Office |
| 3687515 | European Patent Office |
| 3532457 | European Patent Office |
| 3826632 | European Patent Office |
| 3844147 | European Patent Office |
| 3873883 | European Patent Office |
| 3902541 | European Patent Office |
| 4031529 | European Patent Office |
4062910 | European Patent Office |
4138818 | European Patent Office |
4149460 | European Patent Office |
| 4275753 | European Patent Office |
| 40042383 | Hong Kong |
| 40035970 | Hong Kong |
| 40056359 | Hong Kong |
| 40060666 | Hong Kong |
| 40065709 | Hong Kong |
| | | | | |
| Patent Number | Jurisdiction of Filing |
| 40064531 | Hong Kong |
| 40045846 | Hong Kong |
| 40060891 | Hong Kong |
| 40078818 | Hong Kong |
| 40089095 | Hong Kong |
| 507114 | India |
| 528813 | India |
| 570120 | India |
| 292753 | Israel |
| 288617 | Israel |
| 298542 | Israel |
298754 | Israel |
298129 | Israel |
298541 | Israel |
| 7288154 | Japan |
| 7422474 | Japan |
| 7423131 | Japan |
| 7422473 | Japan |
| 7523474 | Japan |
| 7579888 | Japan |
| 7748437 | Japan |
| 7834754 | Japan |
| ZL202180044031.5 | Macao |
| ZL202080087092.5 | Macao |
| ZL202180046463.X | Macao |
| ZL202080050439.9 | Macao |
| ZL202180090269.1 | Macao |
| 404310 | Mexico |
| 411316 | Mexico |
| 412331 | Mexico |
| 415678 | Mexico |
| 788543 | New Zealand |
| 794833 | New Zealand |
| 793361 | New Zealand |
| 794813 | New Zealand |
783166 | New Zealand |
| 2589605 | Republic of Korea |
| 2636385 | Republic of Korea |
| 2023/01086 | South Africa |
2024/03906 | South Africa |
I891942 | Taiwan |
| I860478 | Taiwan |
2585978 | United Kingdom |
| 2586940 | United Kingdom |
| 2592822 | United Kingdom |
| 2595776 | United Kingdom |
| 11,377,416 | United States |
| 11,771,681 | United States |
| 11,773,062 | United States |
| | | | | |
| Patent Number | Jurisdiction of Filing |
| 11,643,390 | United States |
| 11,471,417 | United States |
| 11,406,619 | United States |
| 11,697,638 | United States |
| 11,660,289 | United States |
| 11,578,039 | United States |
| 12,042,564 | United States |
| 12,076,311 | United States |
| 12,084,417 | United States |
| 12,157,723 | United States |
| 12,251,371 | United States |
| 12,318,477 | United States |
| 12,343,327 | United States |
| 12,521,370 | United States |
| 12,649,718 | United States |
In addition, Helus Pharma has title to three provisional patent applications, thirty four US non-provisional patent applications, two hundred and fifty four national (non-US) patent applications, and three Patent Cooperation Treaty (“PCT”) applications, including claims directed to compositions of matter and methods of use in support of its research and development and preclinical and clinical trial programs.
| | | | | | | | |
Patent Application Number | Jurisdiction of Filing | Status |
| 18/027,810 | United States | Pending |
| 18/547,100 | United States | Pending |
| 18/561,152 | United States | Pending |
| 18/576,487 | United States | Pending |
| 18/588,132 | United States | Pending |
| 18/688,125 | United States | Pending |
| 18/707,825 | United States | Pending |
| 18/720,922 | United States | Pending |
| PCT/EP2024/067458 | WIPO | Pending |
| 18/730,397 | United States | Pending |
| 18/730,423 | United States | Pending |
18/825,122 | United States | Pending |
| 18/883,262 | United States | Pending |
| 18/850,356 | United States | Pending |
| 18/852,115 | United States | Pending |
18/867,231 | United States | Pending |
19/020,095 | United States | Pending |
| 19/106,551 | United States | Pending |
| 19/119,308 | United States | Pending |
| 19/170,327 | United States | Pending |
19/159,085 | United States | Pending |
19/481,242 | United States | Pending |
19/488,218 | United States | Pending |
63/928,477 | United States | Pending |
| 19/530,573 | United States | Pending |
| 19/629,961 | United States | Pending |
| PCT/EP2026/067165 | WIPO | Pending |
| | | | | | | | |
Patent Application Number | Jurisdiction of Filing | Status |
| 793553 | New Zealand | Pending |
| 812214 | New Zealand | Pending |
| 832747 | New Zealand | Pending |
| 297492 | Israel | Pending |
325449 | Israel | Pending |
| 3177454 | Canada | Pending |
| NC2022/0016662 | Colombia | Pending |
| NC2026/0005090 | Colombia | Pending |
| NC2026/0005091 | Colombia | Pending |
| MX/a/2022/014605 | Mexico | Pending |
| MX/a/2024/006467 | Mexico | Pending |
| 202203191 | Chile | Pending |
| 10-2022-7040243 | Republic of Korea | Pending |
| 10-2024-7019118 | Republic of Korea | Pending |
| EP21808464.8 | European Patent Office | Pending |
| 24175524.8 | European Patent Office | Pending |
| 202180036163.3 | China | Pending |
| 202410728550.9 | China | Pending |
| 1120220235658 | Brazil | Pending |
| 2021276656 | Australia | Pending |
| 2024203974 | Australia | Pending |
| 11202254530T | Singapore | Pending |
| 10202401521X | Singapore | Pending |
| 202213256 | South Africa | Pending |
| 2201007493 | Thailand | Pending |
| 1-2022-553135 | Philippines | Pending |
1-2024-551326 | Philippines | Pending |
| 202227065770 | India | Pending |
2025-181392 | Japan | Pending |
2025-185270 | Japan | Pending |
| 62023078320.6 | Hong Kong | Pending |
42024099806.2 | Hong Kong | Pending |
| 3186357 | Canada | Pending |
| 10-2023-7003815 | Korea | Pending |
| 10-2026-7007984 | Korea | Pending |
| 2025-185299 | Japan | Pending |
| 21766581.9 | European Patent Office | Pending |
| 62023079716.4 | Hong Kong | Pending |
| 3186359 | Canada | Pending |
| 10-2023-7006128 | Korea | Pending |
| 2021328671 | Australia | Pending |
| 2023-512107 | Japan | Pending |
| 2026-037834 | Japan | Pending |
| 21763068.0 | European Patent Office | Pending |
| 62023079718.0 | Hong Kong | Pending |
| 21786852.0 | European Patent Office | Pending |
| 10-2023-7007858 | Korea | Pending |
| 2021354006 | Australia | Pending |
| 2023-519831 | Japan | Pending |
| | | | | | | | |
Patent Application Number | Jurisdiction of Filing | Status |
| 3194558 | Canada | Pending |
| 62023079720.6 | Hong Kong | Pending |
| 802136 | New Zealand | Pending |
| 305457 | Israel | Pending |
| 3212563 | Canada | Pending |
| NC2023/0013714 | Colombia | Pending |
| MX/a/2023/010843 | Mexico | Pending |
| 202302731 | Chile | Pending |
202501220 | Chile | Pending |
| 10-2023-7032581 | Korea | Pending |
| 22716857.2 | European Patent Office | Pending |
| 202280022029.2 | China | Pending |
| 1120230188946 | Brazil | Pending |
| 2022239825 | Australia | Pending |
| 2026202981 | Australia | Pending |
| 11202305618U | Singapore | Pending |
| 202309486 | South Africa | Pending |
| 2301005753 | Thailand | Pending |
| 1-2023-552572 | Philippines | Pending |
| 202327063524 | India | Pending |
| 2023-556906 | Japan | Pending |
| 62024086011.9 | Hong Kong | Pending |
| 2022277515 | Australia | Pending |
| 2026202771 | Australia | Pending |
| 3216799 | Canada | Pending |
| 22729558.1 | European Patent Office | Pending |
| 202327074210 | India | Pending |
| 2023-571283 | Japan | Pending |
| 10-2023-7041239 | Korea | Pending |
| 62024089505.7 | Hong Kong | Pending |
| 2022342266 | Australia | Pending |
| 3231021 | Canada | Pending |
| 22716971.1 | European Patent Office | Pending |
| 2024-515026 | Japan | Pending |
| 10-2024-7008355 | Korea | Pending |
62024094405.3 | Hong Kong | Pending |
| 2022381220 | Australia | Pending |
| 2026204043 | Australia | Pending |
| 1120240088332 | Brazil | Pending |
| 3236624 | Canada | Pending |
| 202280073355.6 | China | Pending |
| 22783493.4 | European Patent Office | Pending |
| 202417038272 | India | Pending |
| 312175 | Israel | Pending |
| 2024-526529 | Japan | Pending |
| 10-2024-7017594 | Korea | Pending |
| 810005 | New Zealand | Pending |
| 1120242311 | Saudi Arabia | Pending |
62024097476.1 | Hong Kong | Pending |
| | | | | | | | |
Patent Application Number | Jurisdiction of Filing | Status |
| 2023207801 | Australia | Pending |
| 1120240139522 | Brazil | Pending |
3259235 | Canada | Pending |
202380016531.7 | China | Pending |
| 23700949.3 | European Patent Office | Pending |
| 313889 | Israel | Pending |
202417058417 | India | Pending |
2024-541809 | Japan | Pending |
10-2024-7026687 | Korea | Pending |
| MX/a/2024/008691 | Mexico | Pending |
| PI2024003160 | Malaysia | Pending |
| 811765 | New Zealand | Pending |
| 1120243886 | Saudi Arabia | Pending |
62024099899.2 | Hong Kong | Pending |
2023222397 | Australia | Pending |
3244275 | Canada | Pending |
23705529.8 | European Patent Office | Pending |
2024-547671 | Japan | Pending |
10-2024-7028837 | Korea | Pending |
62024101488.0 | Hong Kong | Pending |
| 2023222126 | Australia | Pending |
3244130 | Canada | Pending |
23705530.6 | European Patent Office | Pending |
2024-547667 | Japan | Pending |
10-2024-7028843 | Korea | Pending |
62024101490.6 | Hong Kong | Pending |
2023246690 | Australia | Pending |
3246274 | Canada | Pending |
23715813.4 | European Patent Office | Pending |
2024-557460 | Japan | Pending |
10-2024-7032719 | Korea | Pending |
815769 | New Zealand | Pending |
62025103102.2 | Hong Kong | Pending |
2023242469 | Australia | Pending |
3247035 | Canada | Pending |
23717049.3 | European Patent Office | Pending |
2024-557455 | Japan | Pending |
813800 | New Zealand | Pending |
62025103101.4 | Hong Kong | Pending |
2023331937 | Australia | Pending |
3265884 | Canada | Pending |
23764241.8 | European Patent Office | Pending |
| 2025-512678 | Japan | Pending |
| 10-2025-7007259 | Korea | Pending |
62025111889.4 | Hong Kong | Pending |
| 2023366306 | Australia | Pending |
| 3271746 | Canada | Pending |
| 202380075041.4 | China | Pending |
23798713.6 | European Patent Office | Pending |
| | | | | | | | |
Patent Application Number | Jurisdiction of Filing | Status |
| 2025-524528 | Japan | Pending |
10-2025-7014590 | Korea | Pending |
1120253030 | Saudi Arabia | Pending |
62025112197.1 | Hong Kong | Pending |
2024229430 | Australia | Pending |
1120250180169 | Brazil | Pending |
3284066 | Canada | Pending |
202502558 | Chile | Pending |
202480014984.0 | China | Pending |
NC2025/0011490 | Colombia | Pending |
24708160.7 | European Patent Office | Pending |
322138 | Israel | Pending |
2025-549789 | Japan | Pending |
10-2025-7029225 | Korea | Pending |
MX/a/2025/010013 | Mexico | Pending |
PI2025005159 | Malaysia | Pending |
823179 | New Zealand | Pending |
1-2025-552057 | Philippines | Pending |
1120256479 | Saudi Arabia | Pending |
11202504641X | Singapore | Pending |
2501005646 | Thailand | Pending |
2025/06705 | South Africa | Pending |
62026117641.1 | Hong Kong | Pending |
2024270296 | Australia | Pending |
3290612 | Canada | Pending |
202480035033.1 | China | Pending |
24725094.7 | European Patent Office | Pending |
2025-564163 | Japan | Pending |
10-2025-7040256 | Republic of Korea | Pending |
1120258307 | Saudi Arabia | Pending |
| 62026120459.3 | Hong Kong | Pending |
| 1120220089198 | Brazil | Pending |
| 2023-202672 | Japan | Pending |
18/921,515 | United States of America | Pending |
| 2021391581 | Australia | Pending |
| 3203020 | Canada | Pending |
42024091001.8 | Hong Kong | Pending |
| 202317043169 | India | Pending |
| 303288 | Israel | Pending |
| 2023-533243 | Japan | Pending |
| 800961 | New Zealand | Pending |
| 21 816489.5 | European Patent Office | Pending |
| 2022393234 | Australia | Pending |
| 11 2024 009571 1 | Brazil | Pending |
| 3238583 | Canada | Pending |
| 22 818741.5 | European Patent Office | Pending |
62025102900.0 | Hong Kong | Pending |
| 202417045861 | India | Pending |
| 312859 | Israel | Pending |
| | | | | | | | |
Patent Application Number | Jurisdiction of Filing | Status |
| 2024-529536 | Japan | Pending |
| 10-2024-7020007 | Republic of Korea | Pending |
| MX/a/2024/005955 | Mexico | Pending |
| 811102 | New Zealand | Pending |
| 11202403174T | Singapore | Pending |
| PI 2024002791 | Malaysia | Pending |
| 1120242659 | Saudi Arabia | Pending |
| 202401452 | Chile | Pending |
| 2401003192 | Thailand | Pending |
| 12024551165 | Philippines | Pending |
| 18/711,130 | United States of America | Pending |
| 19/192,691 | United States of America | Pending |
| NC2024/0007518 | Colombia | Pending |
| 202280084101.4 | China | Pending |
24194778.7 | European Patent Office | Pending |
| 42025103259.5 | Hong Kong | Pending |
| PI 2023000584 | Malaysia | Pending |
| 11202300697X | Singapore | Pending |
| 19/202,059 | United States of America | Pending |
| 18/619,547 | United States of America | Pending |
2024242138 | Australia | Pending |
| BR1120250210513 | Brazil | Pending |
| 3,285,080 | Canada | Pending |
CN121194779A | China | Pending |
24716712.5 | European Patent Office | Pending |
| 323233 | Israel | Pending |
2025-556812 | Japan | Pending |
| 10-2025-7035981 | Republic of Korea | Pending |
826414 | New Zealand | Pending |
| 19/469,610 | United States of America | Pending |
| 1120257248 | Saudi Arabia | Pending |
| 202517099658 | India | Pending |
| MX/a/2025/011603 | Mexico | Pending |
| P2025-03094 | United Arab Emirates | Pending |
| 22214748.0 | European Patent Office | Pending |
| 202217028688 | India | Pending |
| 21203394.8 | European Patent Office | Pending |
| 1120220245661 | Brazil | Pending |
| 42023070531.1 | Hong Kong | Pending |
| 202217076779 | India | Pending |
| 1120210243330 | Brazil | Pending |
| 122026010760-6 | Brazil | Pending |
| 18/779,611 | United States of America | Pending |
| 2021284861 | Australia | Pending |
| 202217076899 | India | Pending |
| 2023361184 | Australia | Pending |
| 3,270,486 | Canada | Pending |
| TBD | China | Pending |
| 23 790574.0 | European Patent Office | Pending |
| | | | | | | | |
Patent Application Number | Jurisdiction of Filing | Status |
| 2025-521004 | Japan | Pending |
| 10-2025-7015515 | Republic of Korea | Pending |
| 19/119,888 | United States of America | Pending |
| 3118556 | Canada | Pending |
| 202180046533.1 | China | Pending |
| 18/748,483 | United States of America | Pending |
| 24 215587.7 | European Patent Office | Pending |
| 42025109674.9 | Hong Kong | Pending |
| PCT/EP2025/069677 | WIPO | Pending |
| 114126133 | Taiwan | Pending |
| 2024212866 | Australia | Pending |
| 3280177 | Canada | Pending |
| 202480014812.3 | China | Pending |
| 24 702075.3 | European Patent Office | Pending |
| 322260 | Israel | Pending |
| 2025-542221 | Japan | Pending |
10-2025-7027840 | Republic of Korea | Pending |
| 823545 | New Zealand | Pending |
| 19/149,911 | United States of America | Pending |
| 63/957,091 | United States of America | Pending |
| 63/986,857 | United States of America | Pending |
| 64/093,493 | United States of America | Pending |
Helus Pharma’s patent applications cover a wide range of NSA compounds from different classes, including those with targeted structural modifications for improved pharmacokinetic characteristics and safety profiles without altering their receptor binding. The patent applications also cover novel synthetic routes, pharmaceutical formulations, methods of use, and methods of administration.
Additionally, the Company has entered into multiple licensing agreements that provide the Company with additional access to IP, including the acquisition of an exclusive license to a targeted class of tryptamine-based molecules from Mindset. The licensing agreements collectively provide the Company with access to a broad range of preclinical compounds to support its library of NSA derivative drug candidates.
The Company owns registrations or applications for forty-nine trademarks, including HELUS PHARMATM, EMBARKTM, and HELPING MINDS HEALTM.
The Company’s mission is to provide treatments designed to foster durable improvements in mental healthcare by engineering proprietary drug discovery platforms, innovative drug delivery systems, novel formulation approaches and treatment regimens to address the unmet needs of patients across a multitude of mental health issues. As the Company generates new data it will continue to file or acquire additional patent applications throughout the Company’s development program.
Environmental Protections
The Company is committed to minimizing any environmental impact of its operations and operating its business in a way that will foster sustainable use of the world’s natural resources. At this time, the Company’s business does not materially impact environmental conditions. However, prior to commencing any operations that the Company expects to impact environmental conditions, the Company
will establish internal policies to comply with all applicable environmental protection laws and regulations.
The Company does not expect that there will be any financial or operational effects as a result of environmental protection requirements on its capital expenditures, profit or loss, or its competitive positions in the current fiscal year or in future years.
Competitive Conditions
The Company’s proposed development of psychoactive compounds for use in medical research will compete with other entities that are developing or supplying psychoactive compounds for use in medical research, including clinical trials.
The industry within which the Company intends to operate will become intensely competitive in all its phases, and the Company will face intense competition from other companies, some of which can be expected to have more financial resources and retail, formulation, research, processing, and marketing experience than the Company. Although the Company has access to capital, a management team with specialized skills and knowledge, and an IP portfolio that positions it well among its competitors, there can be no assurance that potential competitors of the Company, which may have greater financial, formulation, research, production, sales and marketing experience, and personnel and resources than the Company, are not currently developing, or will not in the future develop, products and strategies that are equally or more effective and/or economical as any products or strategies developed by the Company or which would otherwise render the Company’s business, products and strategies, as applicable, ineffective, or obsolete. Increased competition by larger and better financed competitors could materially and adversely affect the business, financial condition and results of operations of the Company. See “Risk Factors”.
Negative Operating Cash Flow
Since inception, the Company has had negative operating cash flow and incurred losses. The Company’s negative operating cash flow and losses may continue for the foreseeable future. The Company cannot predict when it will reach positive operating cash flow, if ever. Due to the expected continuation of negative operating cash flow, the Company will be reliant on future financings in order to meet its cash needs. There is no assurance that such future financings will be available on acceptable terms or at all. See “Risk Factors”.
RISK FACTORS
There are various risk factors that could cause the Company’s future results to differ materially from those described in this AIF. The risks and uncertainties described below are those the Company currently believes to be material, but they are not the only ones it faces. If any of the following risks, or any other risks and uncertainties that the Company has not yet identified or that it currently considers not to be material, actually occur or become material risks, the Company’s business, financial condition, results of operations and cash flows, and consequently the price of the Common Shares, could be materially and adversely affected. The risks discussed below also include forward-looking statements and the Company’s actual results may differ substantially from those discussed in these forward-looking statements. See “Note Regarding Forward-Looking Statements” in this AIF.
RISKS RELATED TO THE COMPANY’S BUSINESS AND INDUSTRY
Limited Operating History
The Common Shares commenced trading on Cboe Canada on November 10, 2020 on a post-Transaction basis and therefore the Company has a limited operating history as a public company. To operate effectively, the Company will be required to continue to implement changes in certain aspects of its business, improve information systems and develop, manage and train management-level and other employees to comply with ongoing public company requirements. Failure to take such actions, or delay in implementation thereof, could adversely affect the business, financial condition, liquidity and results of operations of the Company and, more specifically, could result in regulatory penalties, market criticism or the imposition of cease trade orders in respect of the Common Shares.
The Company will be subject to all of the business risks and uncertainties associated with any new business enterprise, including the risk that it will not achieve its operating goals. In order for the Company to meet future operating and debt service requirements, it will need to be successful in its growth, marketing and sales efforts. Additionally, where the Company experiences increased production and future sales, its current operational infrastructure may require changes to scale its business efficiently and effectively to keep pace with demand and achieve long-term profitability. If the Company’s products and services are not accepted by new customers, the Company’s operating results may be materially and adversely affected.
Achieving Publicly Announced Milestones
From time to time, the Company may announce the timing of certain events it expects to occur, such as the anticipated timing of results from clinical trials. These statements are forward-looking and are based on the best estimates of management at the time relating to the occurrence of such events. However, the actual timing of such events may differ from what has been publicly disclosed. The timing of events such as initiation or completion of a clinical trial, filing of an application to obtain regulatory approval, or announcement of additional clinical trials for a prescription drug product candidate may ultimately vary from what is publicly disclosed. See “Commercial Scale Product Manufacturing”, “Safety and Efficacy of Products”, “Clinical Testing and Commercializing Product Candidates”, “Completion of Clinical Trials”, and “Nature of Regulatory Approvals” as discussed under this heading “Risk Factors” for further disclosure of risks and events that may affect the timing of certain events the Company may announce.
The Company undertakes no obligation to update or revise any forward-looking information or statements, whether as a result of new information, future events or otherwise, except as otherwise required by-law. Any variation in the timing of previously announced milestones could have a material adverse effect on the Company’s business plan, financial condition or operating results and the trading price of the Common Shares.
Speculative Nature of Investment Risk
An investment in the securities of the Company carries a high degree of risk and should be considered as a speculative investment. The Company has no history of earnings, limited cash reserves, limited operating history, has not paid dividends, and is unlikely to pay dividends in the immediate or near future.
Early Stage of the Industry and Product Development
Given the early stage of its prescription drug product development, the Company can make no assurance that its research and development programs will result in regulatory approval or commercially viable products. To achieve profitable operations, the Company, alone or with others, must successfully develop, gain regulatory approval for, and market its future products. The Company currently has no products that have been approved by Health Canada, the FDA, the MHRA, the EMA, the Pharmaceutical Drugs Directorate (formerly the Therapeutic Drugs Directorate) (the “PDD”) or any similar regulatory authority. To obtain regulatory approvals for its prescription drug product candidates being developed and to achieve commercial success, clinical trials must demonstrate that the prescription drug product candidates are safe for human use and that they demonstrate efficacy.
Many prescription drug product candidates never reach the stage of clinical testing and even those that do have only a small chance of successfully completing clinical development and gaining regulatory approval. Prescription drug product candidates can fail for a number of reasons, including, but not limited to, being unsafe for human use or due to the failure to provide therapeutic benefits equal to or better than the current standard of treatment at the time of testing. Unsatisfactory results obtained from a particular study relating to a research and development program may cause the Company or its collaborators to abandon commitments to that program. Positive results of early preclinical research may not be indicative of the results that will be obtained in later stages of preclinical or clinical research. Similarly, positive results from early-stage clinical trials may not be indicative of favourable outcomes in later-stage clinical trials, and the Company can make no assurance that any future studies, if undertaken, will yield favourable results.
The early stage of the Company’s product development makes it particularly uncertain whether any of its product development efforts will prove to be successful and meet applicable regulatory requirements, and whether any of its prescription drug product candidates will receive the requisite regulatory approvals, be capable of being manufactured at a reasonable cost or be successfully marketed. If the Company is successful in developing its current and future prescription drug product candidates into approved products, it will still experience many potential obstacles, which would affect its ability to successfully market and commercialize such approved products, such as the need to develop or obtain manufacturing, marketing and distribution capabilities, price pressures from third-party payors, or proposed changes in health care systems. If the Company is unable to successfully market and commercialize any of its products, its financial condition and results of operations may be materially and adversely affected.
The Company can make no assurance that any future studies, if undertaken, will yield favorable results. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in later-stage clinical trials after achieving positive results in early-stage development, and the Company cannot be certain that it will not face similar setbacks. These setbacks have been caused by, among other things, preclinical findings made while clinical trials were underway or safety or efficacy observations made in clinical trials, including previously unreported adverse events or latent defects in the manufactured drug product or the formulation or stability thereof. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their prescription drug product candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to obtain Health Canada, FDA or EMA approval. If the Company fails to produce positive results in future clinical trials and other programs, the development timeline and regulatory approval and commercialization prospects for the Company’s leading prescription drug product candidates, and, correspondingly, its business and financial prospects, would be materially adversely affected.
Preclinical testing and clinical trials for the Company’s products may not achieve the desired results. The results of preclinical testing and clinical trials are uncertain. Product approvals are subject to a number of contingencies and may not be obtained in the time expected or at all. The Company’s products may not attract a following among patients, retailers and/or providers. The Company expects to face an inherent risk of exposure to product liability claims, regulatory action and litigation if the products it plans to distribute are alleged to have caused loss or injury. There can be no assurance that the Company will be able to obtain or maintain product liability insurance on acceptable terms or with adequate coverage against potential liabilities.
The Company’s business relies on its ability to access, develop, and sell HLP003, HLP004, HLP005 and any other compounds the Company is developing or may develop. HLP003, HLP004, certain compounds being developed under the HLP005 Program, and other compounds being developed by the Company are controlled substances in many jurisdictions, including in Canada under Schedule III of the Controlled Drugs and Substances Act and in the United States. The Company may face difficulty accessing the public capital markets in Canada or the United States as a result of the response of regulators, stock exchanges, and other market participants to the Company’s development and sale of a controlled substance. The Company may also have limited access to traditional banking services, as well as limited access to debt financing from traditional institutional lenders. The medical efficacy of HLP003, HLP004 and other compounds being developed under the HLP005 Program has not been confirmed and requires further study and scientific rigour.
Regulatory Risks and Uncertainties
In Canada, certain drugs, including HLP003 and HLP004, are classified as Schedule III drugs under the CDSA and as such, medical and recreational use is illegal under Canadian federal laws. In the United States, certain serotonergic agonists, including HLP003 and HLP004, are classified as Schedule I drugs under the CSA and the Controlled Substances Import and Export Act and as such, medical and recreational use is illegal under the U.S. federal laws. Anyone wishing to conduct research on substances listed in Schedule I under the CSA must register with the DEA and obtain DEA approval of the research proposal. The EU member states currently classify HLP003 and HLP004 as a Schedule I substance under the UN 71 and, as such, a licence is required to produce, dispense, import or export any Schedule I substances, but the specific requirements vary from country to country. Currently in the Netherlands, HLP004 is classified as a List 1 Drug under the Dutch Opium Act and, as such, subject to express authorization being obtained, the production, trade and possession of HLP004 are prohibited. In the United Kingdom, HLP003 and HLP004 are controlled as a Class A drug under the MDA and Schedule 1 drug under the MDR. Schedule 1 drugs can only be lawfully manufactured, produced, possessed and supplied under a controlled drugs domestic licence issued by the UK Home Office.
There is no guarantee that HLP003, HLP004, compounds being developed under the HLP005 Program or any other compounds the Company may develop will ever be approved as medicines in any jurisdiction in which the Company operates. All activities involving such substances by or on behalf of the Company are conducted in accordance with applicable federal, provincial, state and local laws. Further, all facilities engaged with such substances by or on behalf of the Company do so under current licences and permits issued by appropriate federal, provincial and local governmental agencies. While the Company is focused on programs using serotonergic agonist compounds, the Company does not have any direct or indirect involvement with the illegal selling, production or distribution of any substances in the jurisdictions in which it operates and does not intend to have any such involvement. However, the laws and regulations generally applicable to the industry in which the Company is involved in may change in ways currently unforeseen. Any amendment to or replacement of existing laws or regulations, including the classification
or re-classification of the substances the Company is developing or working with, which are matters beyond the Company’s control, may cause the Company’s business, financial condition, results of operations and prospects to be adversely affected or may cause the Company to incur significant costs in complying with such changes or it may be unable to comply therewith. A violation of any applicable laws and regulations of the jurisdictions in which the Company operates could result in significant fines, penalties, administrative sanctions, convictions or settlements arising from civil proceedings initiated by either government entities in the jurisdictions in which the Company operates, or private citizens or criminal charges.
The loss of the necessary licences and permits for any of the above scheduled drugs could have an adverse effect on the Company’s operations.
The NSA segment of the drug industry is a fairly new industry and the Company cannot predict the impact of the ever-evolving compliance regime in respect of this industry. Similarly, the Company cannot predict the time required to secure all appropriate regulatory approvals for future products, or the extent of testing and documentation that may, from time to time, be required by governmental authorities. The impact of compliance regimes, any delays in obtaining, or failure to obtain regulatory approvals may significantly delay or impact the development of markets, its business and products, and sales initiatives and could have a material adverse effect on the business, financial condition and operating results of the Company.
The success of the Company’s business is dependent on the reform of controlled substances laws pertaining to HLP003 and HLP004. If controlled substances laws are not favourably reformed in the United States, the UK, the EU, and other global jurisdictions, the commercial opportunity that the Company is pursuing may be highly limited.
The Company makes no medical, treatment or health benefit claims about the Company’s proposed products. The FDA, Health Canada, the EMA or other similar regulatory authorities have not evaluated claims regarding HLP003, HLP004, or compounds being developed under the HLP005 Program. The efficacy of such products have not been confirmed by approved research. There is no assurance that the use of HLP003, HLP004, or compounds being developed under the HLP005 Program can diagnose, treat, cure or prevent any disease or condition. Vigorous scientific research and clinical trials are needed. The Company is currently conducting clinical trials for the use of its proposed products. Any references to quality, consistency, efficacy and safety of potential products do not imply that the Company verified such in clinical trials or that the Company will complete such trials. If the Company cannot obtain the approvals or research necessary to commercialize its business, it may have a material adverse effect on the Company’s performance and operations.
Risks of Operating in Australia and European Countries
The Company is subject to additional risks related to operating in Australia and certain countries in Europe including: (i) differing regulatory requirements in Australia and Europe; (ii) unexpected changes in price and exchange controls and other regulatory requirements; (iii) increased difficulties in managing the logistics and transportation of collecting and shipping patient material; (iv) import and export requirements and restrictions; (v) compliance with tax, employment, immigration and labour laws for employees living or traveling abroad; (vi) foreign taxes, including withholding of payroll taxes; (vii) foreign currency fluctuations, which could result in increased operating expenses, and other obligations incident to doing business in another country; (viii) difficulties staffing and managing foreign operations; (ix) potential liability under the Corruption of Foreign Public Officials Act of Canada or comparable
foreign regulations; (x) challenges enforcing its contractual and intellectual property rights, especially in Australia and those European countries that do not respect and protect intellectual property rights to the same extent as Canada or the United States; (xi) production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and (xii) business interruptions resulting from geo-political actions, including war and terrorism.
These and other risks associated with the Company’s international operations may materially adversely affect its ability to attain or maintain profitable operations.
“Foreign Private Issuer” Status Under the U.S. Securities Laws
The Company is a “foreign private issuer”, under applicable U.S. federal securities laws, and is, therefore, not subject to the same requirements that are imposed upon U.S. domestic issuers by the SEC. Under the Exchange Act, the Company is subject to reporting obligations that, in certain respects, are less detailed and less frequent than those of U.S. domestic reporting companies. As a result, the Company does not file the same reports that a U.S. domestic issuer would file with the SEC, although the Company is required to file with or furnish to the SEC the continuous disclosure documents that it is required to file in Canada under Canadian securities laws. In addition, the Company’s officers, directors, and principal shareholders are exempt from the reporting and short-swing profit recovery provisions of Section 16 of the Exchange Act. Therefore, the Company’s shareholders may not know on as timely a basis when the Company’s officers, directors and principal shareholders purchase or sell Common Shares, as the reporting periods under the corresponding Canadian insider reporting requirements are longer.
As a foreign private issuer, the Company is exempt from the rules and regulations under the Exchange Act related to the furnishing and content of proxy statements. The Company is also exempt from Regulation FD, which prohibits issuers from making selective disclosures of material non-public information. While the Company complies with the corresponding requirements relating to proxy statements and disclosure of material non-public information under Canadian securities laws, these requirements differ from those under the Exchange Act and Regulation FD and shareholders should not expect to receive the same information at the same time as such information is provided by U.S. domestic companies. In addition, the Company may not be required under the Exchange Act to file annual and quarterly reports with the SEC as promptly as U.S. domestic companies whose securities are registered under the Exchange Act.
The Company May Lose “Foreign Private Issuer” Status in the Future
In order to maintain the Company’s current status as a “foreign private issuer”, a majority of the
Common Shares must be either directly or indirectly held of record by non-residents of the United States unless the Company also satisfies one of the additional requirements necessary to preserve this status. The Corporation may in the future lose its foreign private issuer status if a majority of the Common Shares are held of record by United States residents and the Company fails to meet the additional requirements necessary to avoid loss of foreign private issuer status. The regulatory and compliance costs to the Company under U.S. federal securities laws as a U.S. domestic issuer may be significantly more than the costs it incurs as a Canadian foreign private issuer eligible to use the Multijurisdictional Disclosure System established between Canada and the United States (the “MJDS”). If the Company is not a foreign private issuer, it would not be eligible to use the MJDS or other foreign issuer forms and would be required to file periodic and current reports and registration statements on U.S. domestic issuer forms with the SEC, which are more detailed and extensive than the forms available to a foreign private issuer.
Additionally, the Company may lose the ability to rely upon exemptions from Nasdaq corporate governance requirements that are available to foreign private issuers.
Plans for Growth
The Company intends to continue to advance its research and development programs and operations over the next 12 to 24 months. This advancement will place a significant strain on the Company’s management systems and resources. The Company may not be able to implement its business strategy in a rapidly evolving market. In particular, the Company may be required to manage multiple relationships with various strategic industry participants and other third parties, which relationships could be strained. Similarly, an increase in the number of third-party relationships the Company has, may lead to management of the Company being unable to manage growth effectively. The occurrence of such events may result in the Company being unable to successfully identify, manage and exploit existing and potential market opportunities.
Limited Products
The Company will be heavily reliant on the production and distribution of NSAs and related products. If they do not achieve sufficient market acceptance, it will be difficult for the Company to achieve profitability.
The Company’s revenue will be derived almost exclusively from sales of NSA pharmaceutical products, and the Company expects that its NSA pharmaceutical products will account for substantially all of its revenue for the foreseeable future. If the NSA pharmaceutical market segment declines or fails to achieve substantially greater market acceptance than it currently enjoys, the Company will not be able to grow its revenues sufficiently for it to achieve consistent profitability.
Even if products to be distributed by the Company conform to international safety and quality standards, sales could be adversely affected if consumers in target markets lose confidence in the safety, efficacy, and quality of NSA pharmaceutical products. Adverse publicity about serotonergic agonist pharmaceutical products, similar to those that the Company may sell or the NSAs that the Company may sell could discourage consumers from buying products distributed by the Company.
Limited Marketing and Sales Capabilities
The Company will, for the immediate future, have limited marketing and sales capabilities, and there can be no assurance that it will be able to develop or acquire these capabilities at the level needed to produce and deliver for sale, through industry partners, its products in sufficient commercial quantities. Further, there can be no assurance that the Company, either on its own or through arrangements with other industry participants, will be able to develop or acquire such capabilities on a cost-effective basis, or at all. Finally, there can be no assurance that the Company’s industry partners will be able to market or sell the Company’s products in compliance with requisite regulatory protocols or on a cost-effective basis. The Company’s dependence upon third parties for the production, and marketing or sale, as applicable, of the Company’s products could have a material adverse effect on the Company’s business, financial condition and results of operations.
No Assurance of Commercial Success
The successful commercialization of the Company’s products will depend on many factors, including, the Company’s ability to establish and maintain working partnerships with industry participants in order to
market its products, the Company’s ability to supply a sufficient amount of its products to meet market demand, and the number of competitors within each jurisdiction within which the Company may from time to time be engaged. There can be no assurance that the Company or its industry partners will be successful in their respective efforts to develop and implement, or assist the Company in developing and implementing, a commercialization strategy for the Company’s products.
No Profits or Significant Revenues
The Company has no history upon which to evaluate its performance and future prospects. The Company’s proposed operations are subject to all the business risks associated with new enterprises. These include likely fluctuations in operating results as the Company makes significant investments in research, development and product opportunities, and reacts to developments in its market, including purchasing patterns of customers, and the entry of competitors into the market. The Company will only be able to pay dividends on any shares once its directors determine that it is financially able to do so. The Company cannot make any assurance that it will be profitable in the next three years or generate sufficient revenues to pay dividends to the holders of the Common Shares.
Reliance on Third Parties for Clinical Development Activities
The Company relies and will continue to rely on third parties to conduct a significant portion of its preclinical and clinical development activities. For example, clinical development activities include trial design, regulatory submissions, clinical patient recruitment, clinical trial monitoring, clinical data management and analysis, safety monitoring and project management. If there is any dispute or disruption in its relationship with third parties, or if it is unable to provide quality services in a timely manner and at a feasible cost, the Company’s active development programs will face delays. Further, if any of these third parties fails to perform as the Company expects or if their work fails to meet regulatory requirements, the Company’s testing could be delayed, cancelled or rendered ineffective.
Risks Related to Third Party Relationships
The Company intends to enter into strategic alliances with third parties that the Company believes will complement or augment its proposed business or will have a beneficial impact on the Company. Strategic alliances could present unforeseen integration obstacles or costs, may not enhance the Company’s business, and may involve risks that could adversely affect the Company, including significant amounts of management time that may be diverted from operations in order to pursue and complete such transactions or maintain such strategic alliances. Future strategic alliances could result in the incurrence of additional debt, costs and contingent liabilities, and there can be no assurance that future strategic alliances will achieve, or that the Company’s existing strategic alliances will continue to achieve, the expected benefits to the Company’s business or that the Company will be able to consummate future strategic alliances on satisfactory terms, or at all. Any of the foregoing could have a material adverse effect on the Company’s business, financial condition and results of operations.
In addition to the foregoing, the success of the Company’s business will depend, in large part, on the Company’s ability to enter into, and maintain collaborative arrangements with various participants in the pharmaceutical industry. There can be no assurance that the Company will be able to enter into collaborative arrangements in the future on acceptable terms, if at all. There can be no assurance that such arrangements will be successful, that the parties with which the Company has or may establish arrangements will adequately or successfully perform their obligations under such arrangements, that potential partners will not compete with the Company by seeking or prioritizing alternate, competitor
products. The termination or cancellation of any such collaborative arrangement or the failure of the Company and/or the other parties to these arrangements to fulfill their obligations could have a material adverse effect on the Company’s business, financial condition and results of operations. In addition, disagreements between the Company and any of its industry partners could lead to delays or time consuming and expensive legal proceedings, which could have a material adverse effect on the Company’s business, financial condition and results of operations.
Reliance on Contract Manufacturers
The Company has limited manufacturing experience and relies on contract manufacturing organizations (“CMOs”) to manufacture its prescription drug product candidates for preclinical studies and clinical trials. The Company relies on CMOs for manufacturing, filling, packaging, storing and shipping of drug product in compliance with cGMP regulations applicable to its products. All applicable jurisdictions, including Health Canada, the FDA, the MHRA the EMA, and the PDD, ensure the quality of food, drug products and dietary supplements by carefully monitoring drug manufacturers’ compliance with cGMP regulations. The cGMP regulations for drugs contain minimum requirements for the methods, facilities and controls used in manufacturing, processing and packing of a drug product. There can be no assurances that CMOs will be able to meet the Company’s timetable and requirements. The Company has not contracted with alternate suppliers for drug substance production in the event that the current provider is unable to scale up production, or if it otherwise experiences any other significant problems. If the Company is unable to arrange for alternative third-party manufacturing sources on commercially reasonable terms or in a timely manner, the Company may be delayed in the development of its prescription drug product candidates. Further, CMOs must operate in compliance with cGMP and ensure that their appropriate permits and licences remain in good standing and failure to do so could result in, among other things, the disruption of product supplies. The Company’s dependence upon third parties for the manufacture of its products may adversely affect its profit margins and its ability to develop and deliver products on a timely and competitive basis.
Safety and Efficacy of Products
Before obtaining marketing approval from regulatory authorities for the sale of the Company’s prescription drug product candidates, the Company must conduct preclinical studies in animals and extensive clinical trials in humans to demonstrate the safety and efficacy of the prescription drug product candidates. Clinical testing is expensive and difficult to design and implement, can take many years to complete and has uncertain outcomes. The outcome of preclinical studies and early clinical trials may not predict the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety profiles, notwithstanding promising results in earlier trials. The Company does not know whether the clinical trials it may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market any of its prescription drug product candidates in any jurisdiction. A prescription drug product candidate may fail for safety or efficacy reasons at any stage of the testing process. A major risk the Company faces is the possibility that none of its prescription drug product candidates under development will successfully gain market approval from Health Canada, the FDA, the MHRA, the EMA, the PDD or other regulatory authorities, resulting in the Company being unable to derive any commercial revenue from them after investing significant amounts of capital in their development.
Clinical trials are conducted in representative samples of the potential patient population which may have significant variability. Clinical trials are by design based on a limited number of subjects and of limited
duration for exposure to the product used to determine whether, on a potentially statistically significant basis, the planned safety and efficacy of any such product can be achieved. As with the results of any statistical sampling, the Company cannot be sure that all side effects of its products may be uncovered, and it may be the case that only with a significantly larger number of patients exposed to such product for a longer duration, may a more complete safety profile be identified. Further, even larger clinical trials may not identify rare serious adverse effects, or the duration of such studies may not be sufficient to identify when those events may occur. There have been products that have been approved by the regulatory authorities but for which safety concerns have been uncovered following approval. Such safety concerns have led to labelling changes or withdrawal of such products from the market, and the Company’s products may be subject to similar risks. The Company might have to withdraw or recall its products from the marketplace. The Company may also experience a significant drop in the potential future sales of its products if and when regulatory approvals for such products are obtained, experience harm to its reputation in the marketplace or become subject to lawsuits, including class actions. Any of these results could decrease or prevent any sales of the Company’s products, or substantially increase the costs and expenses of commercializing and marketing its products.
Clinical Testing and Commercializing Products
Before obtaining marketing approval from regulatory authorities for the sale of the Company’s prescription drug product candidates, it must conduct pre-clinical studies in animals and extensive clinical trials in humans to demonstrate the safety and efficacy of the prescription drug product candidates. Clinical testing is expensive and difficult to design and implement, can take many years to complete and has uncertain outcomes. The outcome of pre-clinical studies and early clinical trials may not predict the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety profiles, notwithstanding promising results in earlier trails. The Company does not know whether the clinical trials it may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market any of its prescription drug product candidates in any jurisdiction. A prescription drug product candidate may fail for safety or efficacy reasons at any stage of the testing process. A major risk the Company faces is the possibility that none of its prescription drug product candidates under development will successfully gain market approval from the FDA, or other regulatory authorities, resulting in the Company being unable to derive any commercial revenue from this business segment after investing significant amounts of capital in its development.
The Company cannot predict whether any clinical trials will begin as planned, will need to be restructured, or will be completed on schedule, or at all. The Company’s product development costs will increase if it experiences delays in clinical testing. Significant clinical trial delays could shorten any periods during which the Company may have the exclusive right to commercialize its prescription drug product candidates or allow its competitors to bring products to market before the Company, which would impair the Company’s ability to successfully commercialize its prescription drug product candidates and may harm its financial condition, results of operations and prospects.
The commencement and completion of clinical trials for the Company’s prescription drug product candidates may be delayed for a number of reasons, including but not limited, to:
•failure by regulatory authorities to grant permission to proceed or placing clinical trials on hold;
•suspension or termination of clinical trials by regulators for many reasons, including concerns about patient safety or failure of the Company’s CMOs to comply with cGMP requirements or latent defects in product quality;
•any changes to the Company’s manufacturing process that may be necessary or desired, delays or failure to obtain clinical supply from CMOs of the Company’s products necessary to conduct clinical trials;
•prescription drug product candidates demonstrating a lack of safety or efficacy during clinical trials, reports of clinical testing on similar technologies and products raising safety or efficacy concerns;
•clinical investigators not performing the Company’s clinical trials on their anticipated schedule, dropping out of a trial, or employing methods not consistent with the clinical trial protocol, regulatory requirements or other third parties not performing data collection and analysis in a timely or accurate manner;
•failure of the Company’s contract research organizations to satisfy their contractual duties or meet expected deadlines;
•inspections of clinical trial sites by regulatory authorities;
•regulatory authorities or ethics committees finding regulatory violations that require the Company to undertake corrective action, resulting in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study;
•one or more regulatory authorities or ethics committees rejecting, suspending or terminating the study at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; or
•failure to reach agreement on acceptable terms with prospective clinical trial sites.
The Company’s product development costs will increase if it experiences delays in testing or approval or if the Company needs to perform more or larger clinical trials than planned. Additionally, changes in regulatory requirements and policies may occur, and the Company may need to amend study protocols to reflect these changes. Amendments may require the Company to resubmit its study protocols to regulatory authorities or ethics committees for re-examination, which may impact the cost, timing or successful completion of that trial. Delays or increased product development costs may have a material adverse effect on the Company’s business, financial condition and prospects.
Prior to commencing clinical trials in Canada, the United States, the UK, the Netherlands or other jurisdictions, for any prescription drug product candidates developed by the Company, it may be required to have an IND (or equivalent) for each prescription drug product candidate and to file additional INDs prior to initiating any additional clinical trials. The Company believes that the data from its studies will support the filing of additional INDs to enable the Company to undertake additional clinical studies as it has planned. However, submission of an IND (or equivalent) may not result in the FDA (or equivalent authorities) allowing further clinical trials to begin and, once begun, issues may arise that will require the Company to suspend or terminate such clinical trials.
Additionally, even if relevant regulatory authorities agree with the design and implementation of the clinical trials set forth in an IND, these regulatory authorities may change their requirements in the future. Failure to submit or have effective INDs (or equivalent) and commence or continue clinical programs will significantly limit its opportunity to generate revenue.
Completion of Clinical Trials
As the Company’s prescription drug product candidates advance from preclinical testing to clinical testing, and then through progressively larger and more complex clinical trials, the Company will need to enroll an increasing number of patients that meet its eligibility criteria. There is significant competition for recruiting patients in clinical trials, and the Company may be unable to enroll the patients it needs to complete clinical trials on a timely basis or at all. The factors that affect the Company’s ability to enroll patients are largely uncontrollable and include, but are not limited to, the size and nature of the patient population, eligibility and exclusion criteria for the trial, design of the clinical trial, competition with other companies for clinical sites or patients, perceived risks and benefits of the prescription drug product candidate, and the number, availability, location and accessibility of clinical trial sites.
Commercial Grade Product Manufacturing
The Company’s prescription drug products will be manufactured in small quantities for pre-clinical studies and clinical trials by third party manufacturers. In order to commercialize its product, the Company needs to manufacture commercial quality drug supply for use in registration clinical trials. Most, if not all, of the clinical material used in phase III/pivotal/registration studies must be derived from the defined commercial process including scale, manufacturing site, process controls and batch size. If the Company has not scaled up and validated the commercial production of its product prior to the commencement of pivotal clinical trials, it may have to employ a bridging strategy during the trial to demonstrate equivalency of early-stage material to commercial drug product, or potentially delay the initiation or completion of the trial until drug supply is available. The manufacturing of commercial quality product may have long lead times, may be very expensive and requires significant efforts including, but not limited to, scale-up of production to anticipated commercial scale, process characterization and validation, analytical method validation, identification of critical process parameters and product quality attributes, and multiple process performance and validation runs. If the Company does not have commercial drug supply available when needed for pivotal clinical trials, the Company’s regulatory and commercial progress may be delayed, and it may incur increased product development costs. This may have a material adverse effect on the Company’s business, financial condition and prospects, and may delay marketing of the product.
Nature of Regulatory Approvals
The Company’s development and commercialization activities and prescription drug product candidates are significantly regulated by a number of governmental entities, including Health Canada, the FDA, the MHRA, the EMA and the PDD. Regulatory approvals are required prior to each clinical trial and the Company may fail to obtain the necessary approvals to commence or continue clinical testing. The Company must comply with regulations concerning the manufacture, testing, safety, effectiveness, labeling, documentation, advertising, and sale of products and prescription drug product candidates and ultimately must obtain regulatory approval before it can commercialize a prescription drug product candidate. The time required to obtain approval by such regulatory authorities is unpredictable but typically takes many years following the commencement of preclinical studies and clinical trials. Any analysis of data from clinical activities the Company performs is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. Even if the Company believes results from its sponsored clinical trials are favorable to support the marketing of its prescription drug product candidates, Health Canada, the FDA, the MHRA, the EMA, the PDD or other regulatory authorities may disagree. In addition, approval policies, regulations, or the type and
amount of clinical data necessary to gain approval may change during the course of a prescription drug product candidate’s clinical development and may vary among jurisdictions.
The Company has not obtained regulatory approval for any prescription drug product candidate and it is possible that none of its existing prescription drug product candidates or any future prescription drug product candidates will ever obtain regulatory approval. The Company could fail to receive regulatory approval for its prescription drug product candidates for many reasons, including, but not limited to failure to demonstrate that a prescription drug product candidate is safe and effective for its proposed indication, failure of clinical trials to meet the level of statistical significance required for approval, failure to demonstrate that a prescription drug product candidate’s clinical and other benefits outweigh its safety risks, or deficiencies in the manufacturing processes or the failure of facilities of CMOs with whom the Company contracts for clinical and commercial supplies to pass a pre-approval inspection.
A regulatory authority may require more information, including additional preclinical or clinical data to support approval, which may delay or prevent approval and the Company’s commercialization plans, or the Company may decide to abandon the development program. If the Company were to obtain approval, regulatory authorities may approve any of its prescription drug product candidates for fewer or more limited indications than the Company request, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a prescription drug product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that prescription drug product candidate. Moreover, depending on any safety issues associated with the Company’s prescription drug product candidates that garner approval, Health Canada, the FDA, the MHRA, the EMA, the PDD or other regulatory authorities may impose a risk evaluation and mitigation strategy, thereby imposing certain restrictions on the sale and marketability of such products.
If there are changes in the application of legislation, regulations or regulatory policies, or if problems are discovered with the Company products, or if one of its distributors, licensees or co-marketers fails to comply with regulatory requirements, the regulators could take various actions. These include imposing fines on the Company, imposing restrictions on the Company’s products or its manufacture and requiring the Company to recall or remove its products from the market. The regulators could also suspend or withdraw the Company’s Co-marketing authorizations, requiring it to conduct additional clinical trials, change its labeling or submit additional applications for marketing authorization. If any of these events occurs, the Company’s ability to sell its products may be impaired, and it may incur substantial additional expense to comply with regulatory requirements, which could materially adversely affect its business, financial condition and results of operations.
Market Access and Acceptance
The Company may never have a product that is commercially successful. To date, the Company has no product authorized for marketing. The Company’s future products require further clinical investigation, regulatory review, significant market access and marketing efforts and substantial investment before it can produce any revenue. Furthermore, if approved, the Company’s product may not achieve an adequate level of acceptance by payors, health technology assessment bodies, healthcare professionals, patients and the medical community at large, and the Company may not become profitable. The level of acceptance the Company ultimately achieves may be affected by negative public perceptions and historic media coverage of serotonergic agonist substances, including those resulting in non-ordinary states of consciousness. Because of this history, efforts to educate the medical community and third-party payors and health technologies assessment bodies on the benefits of company’s product compounds may require significant resources and may never be successful, which would prevent the Company from generating
significant revenue or becoming profitable. Market acceptance of the Company’s future products by healthcare professionals, patients, healthcare payors and health technology assessment bodies will depend on a number of factors, many of which are beyond the Company’s control, including, but not limited to, the following:
•acceptance by healthcare professionals, patients and healthcare payors of each product as safe, effective and cost-effective;
•changes in the standard of care for the targeted indications for any product;
•the strength of sales, marketing and distribution support;
•potential product liability claims;
•the product’s relative convenience, ease of use, ease of administration and other perceived advantages over alternatives;
•the prevalence and severity of adverse events or publicity;
•limitations, precautions or warnings listed in the summary of product characteristics, patient information leaflet, package labeling or instructions for use;
•the cost of treatment with the Company’s product in relation to alternatives;
•the steps that prescribers and dispensers must take, as well as the perceived risks based upon its controlled substance status;
•the ability to manufacture the Company’s product in sufficient quantities and yields with adequate purity;
•the availability and amount of coverage and reimbursement from healthcare payors, and the willingness of patients to pay out of pocket in the absence of healthcare payor coverage or adequate reimbursement;
•the willingness of the target patient population to try, and of healthcare professionals to prescribe, the product;
•any potential unfavorable publicity, including negative publicity associated with recreational use or abuse of non-deuterated forms of the Company’s NSAs or other serotonergic agonist compounds; and
•any restrictions on the use, sale or distribution of the Company’s future products.
If the Company’s future products fail to gain market access and acceptance, this will have a material adverse impact on the Company’s ability to generate revenue to provide a satisfactory, or any, return on the Company’s investments. Even if some products achieve market access and acceptance, the market may prove not to be large enough to allow the Company to generate significant revenue.
The Company must rely largely on its own market research to forecast sales as detailed forecasts are not generally obtainable from other sources at this early stage of the industry segment. A failure in the demand for the Company’s products to materialize as a result of competition, technological change or other factors could have a material adverse effect on the business, results of operations and financial condition of the Company.
Unfavourable Publicity or Consumer Perception
The Company believes that the serotonergic agonist pharmaceutical industry segment is highly dependent upon consumer perception regarding the safety, efficacy and quality of the pharmaceutical products. Consumer perception of the Company’s pharmaceutical products can be significantly influenced by scientific research or findings, regulatory investigations, litigation, media attention and other publicity regarding the consumption of serotonergic agonists. There can be no assurance that future scientific
research, findings, regulatory proceedings, litigation, media attention or other research findings or publicity will be favourable to the serotonergic agonist pharmaceutical industry segment or any particular product, or consistent with earlier publicity. Future research reports, findings, regulatory proceedings, litigation, media attention or other publicity that are perceived as less favourable than, or that question, earlier research reports, findings or publicity could have a material adverse effect on the demand for the Company’s NSA products and the business, results of operations, financial condition and cash flows of the Company. The Company’s dependence upon consumer perceptions means that adverse scientific research reports, findings, regulatory proceedings, litigation, media attention or other publicity, whether or not accurate or with merit, could have a material adverse effect on the Company, the demand for the Company’s NSA products, and the business, results of operations, financial condition and cash flows of the Company. Further, adverse publicity reports or other media attention regarding the safety, efficacy and quality of serotonergic agonist products in general, or the Company’s NSA products and services specifically or associating the consumption of serotonergic agonists with illness or other negative effects or events, could have such a material adverse effect. Such adverse publicity reports or other media attention could arise even if the adverse effects associated with such products resulted from consumers’ failure to consume such products legally, appropriately or as directed.
The serotonergic agonist pharmaceutical industry segment is highly dependent upon consumer perception regarding the medical benefits, safety, efficacy and quality of the medicine distributed for medical purposes to such consumers. There can be no assurance that future scientific research or findings on the medical benefits, viability, safety, efficacy and dosing of HLP003, HLP004, or compounds being developed under the HLP005 Program,, regulatory proceedings, litigation, media attention or other research findings or publicity will be favourable to the industry or the Company or any particular product, or consistent with earlier publicity.
Social Media
There has been a marked increase in the use of social media platforms and similar channels that provide individuals with access to a broad audience of consumers and other interested persons. The availability and impact of information on social media platforms is virtually immediate and many social media platforms publish user-generated content without filters or independent verification as to the accuracy of the content posted. Information posted about the Company may be adverse to the Company’s interests or may be inaccurate, each of which may harm the Company’s business, financial condition and results of operations.
Biotechnology and Pharmaceutical Market Competition
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. The Company’s competitors include large, well-established pharmaceutical companies, biotechnology companies, and academic and research institutions developing therapeutics for the same indications the Company is targeting and competitors with existing marketed therapies. Many other companies are developing or commercializing therapies to treat the same diseases or indications for which the Company’s prescription drug product candidates may be useful. Although there are no approved therapies that specifically target opioid addiction, some competitors use therapeutic approaches that may compete directly with the Company’s prescription drug product candidates.
Many of the Company’s competitors have substantially greater financial, technical and human resources than the Company does and have significantly greater experience than the Company in conducting preclinical testing and human clinical trials of product candidates, scaling up manufacturing operations
and obtaining regulatory approvals of products. Accordingly, the Company’s competitors may succeed in obtaining regulatory approval for products more rapidly than the Company does. The Company’s ability to compete successfully will largely depend on:
•the efficacy and safety profile of its prescription drug product candidates relative to marketed products and other prescription drug product candidates in development;
•the Company’s ability to develop and maintain a competitive position in the product categories and technologies on which it focuses;
•the time it takes for the Company’s prescription drug product candidates to complete clinical development and receive marketing approval;
•the Company’s ability to obtain required regulatory approvals;
•the Company’s ability to commercialize any of its prescription drug product candidates that receive regulatory approval;
•the Company’s ability to establish, maintain and protect intellectual property rights related to its prescription drug product candidates; and
•acceptance of any of the Company’s prescription drug product candidates that receive regulatory approval by physicians and other healthcare providers and payers.
Competitors have developed and may develop technologies that could be the basis for products that challenge the discovery research capabilities of prescription drug product candidates the Company is developing. Some of those products may have an entirely different approach or means of accomplishing the same desired therapeutic effect than the Company’s prescription drug product candidates and may be more effective or less costly than its prescription drug product candidates. The success of the Company’s competitors and their products and technologies relative to the Company’s technological capabilities and competitiveness could have a material adverse effect on the future preclinical studies and clinical trials of the Company’s prescription drug product candidates, including its ability to obtain the necessary regulatory approvals for the conduct of such clinical trials. This may further negatively impact the Company’s ability to generate future product development programs using serotonergic agonist based compounds.
If the Company is not able to compete effectively against its current and future competitors, the Company’s business will not grow, and its financial condition and operations will substantially suffer.
Further, there can be no assurance that potential competitors of the Company, which may have greater financial, cultivation, production, sales and marketing experience, and personnel and resources than the Company, are not currently developing, or will not in the future develop, products and strategies that are equally or more effective and/or economical as any products or strategies developed by the Company or which would otherwise render the Company’s business, products and strategies, as applicable, ineffective, or obsolete. Increased competition by larger and better financed competitors could materially and adversely affect the business, financial condition and results of operations of the Company.
Reliance on Key Executives and Scientists
The loss of key members of the Company’s staff, could harm the Company. The Company does not have employment agreements with all members of its staff, although such employment agreements do not guarantee their retention. The Company also depends on its scientific and clinical collaborators and advisors, all of whom have outside commitments that may limit their availability to the Company. In addition, the Company believes that its future success will depend in large part upon its ability to attract and retain highly skilled scientific, managerial, medical, manufacturing, clinical and regulatory personnel, particularly as the Company expands its activities and seeks regulatory approvals for clinical trials. The
Company enters into agreements with its scientific and clinical collaborators and advisors, key opinion leaders and academic partners in the ordinary course of its business. Should key academic and scientific personnel including employees or collaborative partners who work on the development of the Company’s research activities leave, the Company’s current and future development programs may be delayed or adversely affected. Notwithstanding these arrangements, the Company faces significant competition for these types of personnel from other companies, research and academic institutions, government entities and other organizations. The Company cannot predict its success in hiring or retaining the personnel it requires for continued growth. In addition, due to limited financial resources, the Company may not be able to successfully expand its operations due to challenges in recruiting and training qualified new staff. Expansion of personnel may result in significant diversion of management time and resources. The loss of the services of any of the Company’s executive officers or other key personnel could potentially harm its business, operating results or financial condition.
Employee Misconduct
Notwithstanding having established an insider trading policy and code of ethics and business conduct, the Company is exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include failures to comply with Health Canada, the FDA the MHRA the EMA the PDD, and other comparable international authorities’ regulations, provide accurate information to Health Canada, the FDA the MHRA, the EMA, and/or the PDD provide accurate information to Health Canada, the FDA, the MHRA, the EMA and the PDD, comply with manufacturing standards the Company has established, comply with federal and provincial healthcare fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to the Company. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing, and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to the Company’s reputation. If any such actions are instituted against the Company, and the Company is not successful in defending itself or asserting its rights, those actions could have a substantial impact on the Company’s business and results of operations, including the imposition of substantial fines or other sanctions.
Business Expansion and Growth
The Company may in the future seek to expand its pipeline and capabilities by acquiring one or more companies or businesses, entering into collaborations, or in-licensing one or more prescription drug product candidates. Acquisitions, collaborations and in-licences involve numerous risks, including, but not limited to substantial cash expenditures, technology development risks, potentially dilutive issuances of equity securities, incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition, difficulties in assimilating the operations of the acquired companies, entering markets in which the Company has limited or no direct experience, and potential loss of the Company’s key employees or key employees of the acquired companies or businesses.
The Company has experience in making acquisitions, entering collaborations and in-licensing prescription drug product candidates; however, the Company cannot provide assurance that any acquisition, collaboration or in-licence will result in short-term or long-term benefits to it. The Company may incorrectly judge the value or worth of an acquired company or business or in-licensed prescription drug product candidate. In addition, the Company’s future success would depend in part on its ability to
manage the rapid growth associated with some of these acquisitions, collaborations and in-licences. The Company cannot provide assurance that it would be able to successfully combine its business with that of acquired businesses, manage a collaboration or integrate in-licensed prescription drug product candidates. Furthermore, the development or expansion of the Company’s business may require a substantial capital investment by the Company.
Negative Results of External Clinical Trials or Studies
From time to time, studies or clinical trials on various aspects of breakthrough neuropsychiatry products are conducted by academic researchers, competitors or others. The results of these studies or trials, when published, may have a significant effect on the market for the breakthrough neuropsychiatry product that is the subject of the study. The publication of negative results of studies or clinical trials or adverse safety events related to the Company’s prescription drug product candidates, or the therapeutic areas in which the Company’s prescription drug product candidates compete, could adversely affect its share price and the Company’s ability to finance future development of its prescription drug product candidates, and its business and financial results could be materially and adversely affected.
Product Liability
The Company currently does not carry any product liability insurance coverage. Even though the Company is not aware of any product liability claims at this time, its business exposes itself to potential product liability, recalls and other liability risks that are inherent in the sale of consumer products. The Company can provide no assurance that such potential claims will not be asserted against it. A successful liability claim or series of claims brought against the Company could have a material adverse effect on its business, financial condition and results of operations.
Although the Company intends to obtain adequate product liability insurance, it cannot provide any assurances that it will be able to obtain or maintain adequate product liability insurance of on acceptable terms, if at all, or that such insurance will provide adequate coverage against potential liabilities. Claims or losses in excess of any product liability cover that may be obtained by the Company could have a material adverse effect on its business, financial conditional and results of operations.
Some of the Company’s agreements with third parties might require it to maintain product liability insurance. If the Company cannot obtain acceptable amounts of coverage on commercially reasonable terms in accordance with the terms set forth in these agreements, the corresponding agreements would be subject to termination, which could have a material adverse impact on its operations.
Enforcing Contracts
Due to the nature of the business of the Company and the fact that certain of its contracts involve HLP003 and HLP004, the use of which is not legal under Canadian or U.S. federal law and in certain other jurisdictions, the Company may face difficulties in enforcing its contracts in Canadian or U.S. federal and state courts. The inability to enforce any of its contracts could have a material adverse effect on its business, operating results, financial condition or prospects.
In order to manage its contracts with contractors, the Company will ensure that such contractors are appropriately licensed. Were such contractors to operate outside the terms of these licences, the Company may experience an adverse effect on its business, including the pace of development of its product.
Product and Material Recalls
Manufacturers, producers and distributors of products are sometimes subject to the recall or return of their products for a variety of reasons, including product defects, such as contamination, unintended harmful side effects or interactions with other substances, packaging safety storage deficiencies and inadequate or inaccurate labelling disclosure. If any of the Company’s products are recalled due to an alleged product defect or for any other reason, the Company could be required to incur the unexpected expense of the recall and any legal proceedings that might arise in connection with the recall. The Company may have to recall material being used in a clinical trial resulting in delays to the trial and additional manufacturing expenses, if further drug product is required. If the product is already commercialized, the Company may lose a significant amount of sales and may not be able to replace those sales at an acceptable margin or at all. In addition, a product recall may require significant management attention.
Although the Company’s suppliers have detailed procedures in place for testing its products, there can be no assurance that any quality, potency or contamination problems will be detected in time to avoid unforeseen product recalls, regulatory action or lawsuits. Additionally, if the Company is subject to recall, the image of the Company could be harmed. A recall for any of the foregoing reasons could lead to decreased demand for the Company’s products and could have a material adverse effect on the results of operations and financial condition of the Company. Additionally, product recalls may lead to increased scrutiny of the Company’s operations by regulatory agencies, requiring further management attention, potential loss of applicable licences and potential legal fees and other expenses.
Distribution and Supply Chain Interruption
The Company is susceptible to risks relating to distributor and supply chain interruptions. Distribution in the U.S., Canada, the EU, the UK and other jurisdictions will be largely accomplished through independent contractors, therefore, an interruption (e.g., a labour strike) for any length of time affecting such independent contractors may have a significant impact on the Company’s ability to sell or manufacture its products. Supply chain interruptions, including a production or inventory disruption, could impact product quality and availability. Inherent to producing products is a potential for shortages or surpluses in future years if demand and supply are materially different from long-term forecasts. The Company monitors category trends and regularly reviews maturing inventory levels.
Difficulty to Forecast
The Company must rely largely on its own market research to forecast sales as detailed forecasts are not generally obtainable from other sources at this early stage of the serotonergic agonist pharmaceutical industry segment. A failure in the demand for the Company’s NSA pharmaceutical industry products to materialize as a result of competition, technological change or other factors could have a material adverse effect on the business, results of operations and financial condition of the Company.
Promoting the Brand
Promoting the Company’s brand will be critical to creating and expanding a customer base. Promoting the brand will depend largely on the Company’s ability to provide NSA pharmaceutical products to the market. Further, the Company may, in the future, introduce new products or services that its customers do not like, which may negatively affect the brand and reputation. If the Company fails to successfully
promote its brand or if it incurs excessive expenses in this effort, its business and financial results from operations could be materially adversely affected.
If there are changes in the applicable regulatory framework governing the promotion, branding and marketing of the Company’s products, the Company’s ability to promote and sell its products may be impaired, and it may incur substantial additional expense to comply with regulatory requirements, which could materially adversely affect its business, financial condition and results of operations.
Product Viability
If the Company’s pharmaceutical products are not perceived to have the effects intended by the end user, the Company’s business may suffer. In general, pharmaceutical products in the class of drugs including HLP003 and HLP004 have minimal long-term data with respect to efficacy, unknown side effects and/or interaction with individual human biochemistry or other supplements or medications. As a result, the Company’s pharmaceutical products could have certain side effects if not used as directed or if taken by an end user that has certain known or unknown medical conditions.
Success of Quality Control Systems
The quality and safety of the Company’s products are critical to the success of its business and operations. As such, it is imperative that the Company (and its service providers’) quality control systems operate effectively and successfully. Quality control systems can be negatively impacted by the design of the quality control systems, the quality of training programs and adherence by employees to quality control guidelines. Any significant failure or deterioration of such quality control systems could have a material adverse effect on the Company’s business and operating results.
Reliance on Key Inputs
The Company’s business is expected to be dependent on a number of key inputs and their related costs including raw materials and supplies. Any significant interruption or negative change in the availability or economics of the supply chain for key inputs could materially impact the business, financial condition and operating results of the Company. Any inability to secure required supplies and services or to do so on appropriate terms could have a materially adverse impact on the business, financial condition and operating results of the Company.
Liability Arising from Fraudulent or Illegal Activity
The Company is exposed to the risk that its employees, independent contractors, consultants, service providers and licensors may engage in fraudulent or other illegal activity. Misconduct by these parties could include intentional undertakings of unauthorized activities, or reckless or negligent undertakings of authorized activities, in each case on the Company’s behalf or in its service that violate (i) various laws and regulations, including healthcare laws and regulations, (ii) laws that require the true, complete and accurate reporting of financial information or data, (iii) the terms of the Company’s agreements with third parties. Such misconduct could expose the Company to, among other things, class actions and other litigation, increased regulatory inspections and related sanctions, and lost sales and revenue or reputational damage.
The precautions taken by the Company to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting the Company from governmental
investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Such misconduct may result in legal action, significant fines or other sanctions and could result in loss of any regulatory licence held by the Company at such time. The Company may be subject to security breaches at its facilities or in respect of electronic document or data storage, which could lead to breaches of applicable privacy laws and associated sanctions or civil or criminal penalties; events, including those beyond the control of the Company, may damage its operations. In addition, these events may negatively affect customers’ demand for the Company’s products. Such events include, but are not limited to, non-performance by third party contractors; increases in materials or labour costs; breakdown or failure of equipment; failure of quality control processes; contractor or operator errors; and major incidents and/or catastrophic events such as fires, explosions, earthquakes or storms. As a result, there is a risk that the Company may not have the capacity to meet customer demand or to meet future demand when it arises. Failure to comply with health and safety laws and regulations may result in additional costs for corrective measures, penalties or in restrictions on the Company’s manufacturing operations.
Operating Risk and Insurance Coverage
The Company has directors and officers insurance to protect its assets, operations and employees. The Company’s insurance is subject to coverage limits and exclusions and may not be available for the risks and hazards to which the Company is expected to be exposed. In addition, no assurance can be given that such insurance will be adequate to cover the Company’s liabilities or will be generally available in the future, or if available, that premiums will be commercially justifiable. If the Company were to incur substantial liability and such damages were not covered by insurance or were in excess of policy limits, or if the Company were to incur such liability at a time when it is not able to obtain liability insurance, its business, results of operations and financial condition could be materially adversely affected.
Costs of Operating as Public Company
As a public company, the Company will incur significant legal, accounting and other expenses. As a public company, the Company is subject to various securities rules and regulations, which impose various requirements on the Company, including the requirement to establish and maintain effective disclosure and financial controls and corporate governance practices. The Company’s management and other personnel need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase the Company’s legal and financial compliance costs and make some activities more time-consuming and costly.
Management of Growth
The Company may be subject to growth-related risks, including capacity constraints and pressure on its internal systems and controls. The ability of the Company to manage growth effectively will require it to continue to implement and improve its operational and financial systems and to expand, train and manage its employee base. The inability of the Company to deal with this growth may have a material adverse effect on the Company’s business, financial condition, results of operations and prospects.
Conflicts of Interest
The Company may be subject to various potential conflicts of interest because of the fact that some of its officers and directors may be engaged in a range of business activities. The Company’s executive officers and directors may devote time to their outside business interests, so long as such activities do not materially or adversely interfere with their duties to the Company. In some cases, the Company’s
executive officers and directors may have fiduciary obligations associated with these business interests that interfere with their ability to devote time to the Company’s business and affairs and that could adversely affect the Company’s operations. These outside business interests could require significant time and attention of the Company’s executive officers and directors.
In addition, the Company may also become involved in other transactions which conflict with the interests of its directors and the officers who may from time-to-time deal with persons, firms, institutions or companies with which the Company may be dealing, or which may be seeking investments similar to those desired by it. The interests of these persons could conflict with those of the Company, and from time to time, these persons may be competing with the Company for available investment opportunities.
Conflicts of interest, if any, will be subject to the procedures and remedies provided under applicable laws. In particular, in the event that such a conflict of interest arises at a meeting of the Company’s directors, a director who has such a conflict will abstain from voting for or against the approval of such participation or such terms. In accordance with applicable laws, the Board is required to act honestly, in good faith and in the best interests of the Company.
Foreign Operations
In addition to operations carried out in Canada and the UK, the Company intends to carry out international operations through an office in Ireland. As a result, the Company may be subject to political, economic and other uncertainties, including, but not limited to, additional implications that may have a material impact on the Company’s ability to operate in other jurisdictions including:
•differences in the regulatory requirements for drug approvals;
•differing requirements for securing, maintaining or obtaining freedom to operate;
•the potential for reduced protection for intellectual property rights;
•challenges with compliance to different regulations and court systems of multiple jurisdictions and
•compliance with a wide variety of foreign laws, treaties and regulations;
•differing reimbursement regimes and price controls in certain international markets;
•differing labor relations that create challenges with staffing and managing international operations; and
•impacts on manufacturing capabilities leading to production shortages.
The Company’s international operations may also be adversely affected by laws and policies of Canada affecting foreign trade, taxation and investment. In the event of a dispute arising in connection with its foreign operations, the Company may be subject to the exclusive jurisdiction of foreign courts or may not be successful in subjecting foreign persons to the jurisdiction of courts in Canada or enforcing Canadian judgments in foreign jurisdictions.
Similarly, to the extent that the Company’s assets are located outside of Canada, investors may have difficulty collecting from the Company any judgments obtained in the Canadian courts and predicated on the civil liability provisions of securities laws. Consequently, investors may be effectively prevented from pursuing remedies against the Company under Canadian securities laws or otherwise. The Company may also be hindered or prevented from enforcing its rights with respect to a governmental entity or instrumentality because of the doctrine of sovereign immunity.
Exchange Rate Fluctuations
Due to the international scope of the Company’s current and future operations, the Company’s assets, future earnings and cash flows may be influenced by movements in exchange rates of several currencies, particularly the British Pound, the United States dollar, Canadian Dollar and the Euro. The Company’s reporting currency is denominated in United States dollars and the Company’s functional currency is the United States dollar and the majority of the Company’s operating expenses are paid in United States dollars. The Company may also regularly acquire services, consumables and materials in British Pounds, United States dollars, Canadian dollars and other currencies. Further, future revenue may be derived from abroad. As a result, the Company’s business and the price of the Company’s products may be affected by fluctuations in foreign exchange rates between the British Pound, the United States dollar, the Canadian dollar and other currencies, which may also have a significant impact on the Company’s results of operations and cash flows from period to period. Currently, the Company does not have any exchange rate hedging arrangements in place.
Cybersecurity and Privacy Risk
The Company’s information systems and any third-party service providers and vendors are vulnerable to an increasing threat of continually evolving cybersecurity risks. These risks may take the form of malware, computer viruses, cyber threats, extortion, employee error, malfeasance, system errors or other types of risks, and may occur from inside or outside of the respective organizations. Cybersecurity risk is increasingly difficult to identify and quantify and cannot be fully mitigated because of the rapidly evolving nature of the threats, targets and consequences. Additionally, unauthorized parties may attempt to gain access to these systems through fraud or other means of deceiving third-party service providers, employees or vendors. The Company’s operations depend, in part, on how well networks, equipment, IT systems and software are protected against damage from a number of threats. These operations also depend on the timely maintenance, upgrade and replacement of networks, equipment, IT systems and software, as well as pre-emptive expenses to mitigate the risks of failures. However, if the Company is unable or delayed in maintaining, upgrading or replacing IT systems and software, the risk of a cybersecurity incident could materially increase. Any of these and other events could result in information system failures, delays and/or increases in capital expenses. The failure of information systems or a component of information systems could, depending on the nature of any such failure, adversely impact the Company’s reputation and results of operations.
The Company may collect and store certain personal information about customers and are responsible for protecting such information from privacy breaches. A privacy breach may occur through procedural or process failure, information technology malfunction, or deliberate unauthorized intrusions. In addition, theft of data is an ongoing risk whether perpetrated via employee collusion or negligence or through deliberate cyber-attack. Any such privacy breach or theft could have a material adverse effect on the Company’s business, financial condition and results of operations.
In addition, there are a number of laws protecting the confidentiality of certain patient health information, including patient records, and restricting the use and disclosure of that protected information. In particular, the privacy rules under the Personal Information Protection and Electronics Documents Act (Canada) (“PIPEDA”) and where applicable, provincial legislation governing personal health information, protect medical records and other personal health information by limiting their use and disclosure of health information to the minimum level reasonably necessary to accomplish the intended purpose. If the Company were found to be in violation of the privacy or security rules under PIPEDA or other laws protecting the confidentiality of medical patients health information, the Company could be subject to sanctions and civil or criminal penalties, which could increase its liabilities, harm its reputation
and have a material adverse effect on the Company’s business, financial condition and results of operations.
Risks Related to Artificial Intelligence
The Company uses, and may further adopt, artificial intelligence (“AI”) technologies to support research, development, data analysis, and operational activities. The use of AI involves risks, including potential errors, biases, or limitations in AI outputs and the underlying data sets, which could adversely affect research results, development decisions, or regulatory submissions. The regulatory framework governing AI, particularly in healthcare and life sciences, is evolving and may impose additional compliance requirements or restrict certain AI applications, increasing costs or delaying development. The Company may also rely on third-party AI tools, which may present risks related to data security, intellectual property rights, and service availability. Any failure to effectively manage these risks could have a material adverse effect on the Company’s business and prospects.
Environmental Regulation and Risks
The Company’s operations are subject to environmental regulations that mandate, among other things, the maintenance of air and water quality standards and land reclamation. They also set forth limitations on the generation, transportation, storage and disposal of solid and hazardous waste. Environmental legislation is evolving in a manner which could include stricter standards and enforcement, increased fines and penalties for non-compliance, more stringent environmental assessments of proposed projects and a heightened degree of responsibility for companies and their officers, directors and employees. There is no assurance that future changes in environmental regulation, if any, will not adversely affect the Company’s operations.
Failure to comply with applicable laws, regulations and permitting requirements may result in enforcement actions thereunder, including orders issued by regulatory or judicial authorities causing operations to cease or be curtailed, and may include corrective measures requiring capital expenditures, installation of additional equipment, or remedial actions. The Company may be required to compensate those suffering loss or damage by reason of its operations and may have civil or criminal fines or penalties imposed for violations of applicable laws or regulations.
Amendments to current laws, regulations and permits governing NSAs and related products, or more stringent implementation thereof, could have a material adverse impact on the Company and cause increases in expenses, capital expenditures or production costs or reduction in levels of production or require abandonment or delays in development.
Legalization of Scheduled Serotonergic Agonists
Despite the current status of many psychotropic substances as a Schedule II and Schedule I controlled substances in the United States and Canada, respectively, there may be changes in the status of some of these substances under the laws of certain jurisdictions. The legalization of scheduled serotonergic agonists with inadequate regulatory oversight may lead to the development of psychotropic tourism in such states in clinics without proper therapeutic infrastructure or adequate clinical research. The expansion of such an industry which could put patients at risk may bring reputational and regulatory risk to the entire serotonergic agonist industry, leading to challenges for the Company to achieve regulatory approval. The legalization of psychotropic compounds in the future may also impact commercial sales for
the Company due to a reduced barrier to entry for serotonergic agonists leading to a risk of increasing competition.
Forward-looking statements May Prove to be Inaccurate
Investors should not place undue reliance on forward-looking statements. By their nature, forward-looking statements involve numerous assumptions, known and unknown risks and uncertainties, of both general and specific nature, that could cause actual results to differ materially from those suggested by the forward-looking statements or contribute to the possibility that predictions, forecasts or projections will prove to be materially inaccurate.
Effects of Inflation
Global markets have experienced increased rates of inflation. Inflation itself, as well as certain governmental efforts to combat inflation, may have significant negative effects on any economy which the Company does business. Past governmental efforts to curb inflation have involved certain drastic economic measures, which had a materially adverse impact on the level of economic activity in these countries. Any future economic measures to curb inflation could be expected to have similar adverse effects on the level of economic activity in the market which the Company does business and, in turn, on the operations of the Company.
Political and Economic Conditions
Political and economic conditions directly affect the Company’s business and can result in a material adverse effect on the Company. Macroeconomic policies imposed by foreign governments could have significant impact on the Company. As certain global markets experience increased inflation, certain government actions to control inflation may have significant impact on the Company.
The Company cannot control or predict foreign government implementation of changes to existing policies that may impact the Company’s operations in foreign markets and, consequently, its business. The Company’s business, operating results and financial condition and prospects, as well as the market price of its securities, may be adversely affected by changes in government public policies, whether federal, state or local, that affect, without limitation:
•inflation;
•fluctuations in exchange rates;
•exchange controls and restrictions on remittances abroad;
•interest rates and monetary policies;
•import and export controls;
•liquidity of domestic capital, credit and financial markets;
•expansion or contraction of foreign economies, as measured by rates of growth in gross domestic product;
•fiscal policies; and
•other political, social and economic developments in or affecting foreign markets.
Government policies and measures to combat inflation, along with public speculation about such policies and measures, have often had adverse effects on global economies, have contributed to economic uncertainty and may increase volatility in foreign securities markets. Government action to control
inflation may involve actions such as price and salary controls, currency devaluations, capital limitations, limits on imports and other actions which could significantly impact the operations of the Company.
Other policies and measures adopted by governments, include interest rate adjustments, intervention in the currency markets or actions to adjust or fix the value of the local currency may adversely affect the target market’s economy, the Company’s business and results of operations.
Uncertainty over whether federal governments will implement reforms or changes in policy or regulation affecting these or other factors in the future may affect economic performance and contribute to economic uncertainty in markets that the Company operates or relies on, which may in turn have adverse effects on the Company’s operations in the market and consequently on the results of its operations.
Litigation Risk
As of the date hereof, no litigation or class proceedings have been commenced or certified. Should any litigation or class actions that the Company becomes involved in be unable to be resolved favourably or if any claims or litigation are determined against the Company, the Company’s financial position, operating results and the trading price of the Common Shares could be materially adversely affected.
The Company faces the risk of various claims, legal proceedings, and class actions. Such actions could result in liabilities or necessitate operational changes, negatively impacting results. Even favorable resolutions can divert resources, generate substantial legal costs, and harm reputation.
The Company may be exposed to securities class action investigations or proceedings, especially following share price volatility or public disclosures. These proceedings could allege violations of securities laws or misrepresentations. Defending such claims are potentially costly and time-consuming and may lead to significant monetary judgments, settlements, or regulatory penalties. Any liability exceeding insurance coverage could materially and adversely affect the Company’s business, financial condition, and results.
Application and Interpretation of Tax Laws
The Company is subject to direct and indirect taxes in various foreign jurisdictions. The amount of tax that the Company pays, directly or indirectly, is subject to the interpretation of applicable tax laws in the jurisdictions of operations in which the Company has interests. The Company has taken and will continue to take tax positions based on the application and interpretation of tax laws, but tax accounting often involves complex matters and judgment is required in determining the Company’s foreign provisions for taxes and other tax liabilities. There can be no assurance that a taxing authority will not have a different interpretation of the law and assess the Company, or the operations in which the Company has interests, with additional taxes. Further, the Company’s future effective tax rates could be impacted by changes in tax laws or regulations, and changing interpretation of existing laws or regulations. Both domestic and international tax laws, and interpretation of the tax laws, are subject to change as a result of changes in fiscal policy, changes in legislation, evolution of regulation and court rulings. The application of these tax laws and related regulations is subject to legal and factual interpretation, judgment and uncertainty.
Enforcement of Civil Liabilities
Certain of the Company’s subsidiaries and assets are located outside of Canada. Accordingly, it may be difficult for investors to enforce within Canada any judgments obtained against the Company, including
judgments predicated upon the civil liability provisions of applicable Canadian securities laws or otherwise. Consequently, investors may be effectively prevented from pursuing remedies against the Company under Canadian securities laws or otherwise.
The Company has subsidiaries incorporated in the United States and Ireland. It may not be possible for shareholders to effect service of process outside of Canada against the directors and officers of the Company who are not resident in Canada. In the event a judgment is obtained in a Canadian court against one or more of such persons for violations of Canadian securities laws or otherwise, it may not be possible to enforce such judgment against persons not resident in Canada. Additionally, it may be difficult for an investor, or any other person or entity, to assert Canadian securities law or other claims in original actions instituted in the United States and Ireland. Courts in such jurisdictions may refuse to hear a claim based on a violation of Canadian securities laws or otherwise on the grounds that such jurisdiction is not the most appropriate forum to bring such a claim. Even if a foreign court agrees to hear a claim, it may determine that the local law, and not Canadian law, is applicable to the claim. If Canadian law is found to be applicable, the content of applicable Canadian law must be proven as a fact, which can be a time-consuming and costly process. Certain matters of procedure will also be governed by foreign law.
Pandemics, Epidemics and Other Health Risks
Pandemics, epidemics and other health risks could have an adverse effect on the Company’s business. Pandemics, epidemics and other health risks could occur, which could adversely affect the Company’s ability to conduct its operations as currently conducted, or the ability of suppliers to provide the Company with products and services needed to operate the business.
Pandemics, epidemics and other health risks could have an adverse effect on the economy and financial markets, resulting in a decline of commercial activity. Any of these events could have an adverse effect on the Company’s business and financial performance.
RISKS RELATED TO INTELLECTUAL PROPERTY
Trademark Protection
Failure to register or maintain trademarks for the Company or its products could require the Company to rebrand its products resulting in a material adverse impact on its business.
Trade Secrets
The Company relies on third parties to develop its products and as a result, must share trade secrets with them. The Company seeks to protect its proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with its collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically restrict the ability of the Company’s collaborators, advisors, employees and consultants to publish data potentially relating to its trade secrets. Its academic and clinical collaborators typically have rights to publish data, provided that the Company is notified in advance and may delay publication for a specified time in order to secure any intellectual property rights arising from the collaboration. In other cases, publication rights are controlled exclusively by the Company, although in some cases the Company may share these rights with other parties. The Company may also conduct joint research and development programs which may require it to share trade secrets under the terms of research and development collaboration or similar agreements. Despite the Company’s efforts to protect its trade secrets, the Company’s competitors may
discover its trade secrets, either through breach of these agreements, independent development or publication of information. A competitor’s discovery of the Company’s trade secrets may impair its competitive position and could have a material adverse effect on its business and financial condition.
Patent Law Reform
The Company’s commercial success depends in part on obtaining and maintaining patents and other forms of intellectual property rights for its current and future therapeutic candidates and associated therapies, digital therapies, methods used to manufacture the underlying therapeutic substances, and the methods for treating patients using those substances and therapies, or on licensing in such rights. Failure to obtain, maintain, protect, enforce or extend adequate patent and other intellectual property rights could materially adversely affect the Company’s ability to develop and market its current and future therapeutic candidates. The Company also relies on trade secrets and know-how to develop and maintain its proprietary and intellectual property position. Any failure to protect its trade secrets and know-how could adversely affect the Company’s operations and prospects.
The Company cannot be certain that patents will be issued or granted with respect to patent applications that are currently pending, or that issued or granted patents will not later be found to be invalid or unenforceable. The patent position of companies like the Company is generally uncertain because it involves complex legal and factual considerations. The standards applied by the UK Intellectual Property Office, the European Patent Office, the USPTO, the Canadian Intellectual Property Office (the “CIPO”) and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in pharmaceutical patents. Consequently, patents may not issue from the Company’s pending patent applications, and even if they do issue, such patents may not issue in a form that effectively prevents others from developing or commercializing competing therapies. As such, the Company does not know the degree of future protection that it will have on its proprietary therapies.
The patent prosecution process is expensive, complex and time-consuming, and the Company, its current or future third-party partners, licensors, licensees, or collaboration partners may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that the Company or its licensors, licensees or collaboration partners will fail to identify patentable aspects of inventions made in the course of research, development or commercialization activities before it is too late to pursue patent protection on them. In addition, although the Company enters into non-disclosure and confidentiality agreements with parties who have access to confidential or patentable aspects of its R&D output, such as its employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors, and other third parties, any of these parties may breach the agreements and disclose such output before a patent application is filed, thereby jeopardizing the Company’s ability to seek patent protection. Furthermore, publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the UK and other jurisdictions are typically not published until 18 months after filing, or in some cases not published until and unless granted. Therefore, the Company cannot be certain that it is the first to make the inventions claimed in its patents or pending patent applications, or that it was the first to file for patent protection of such inventions. Similarly, the Company cannot be certain that for any licensed patents or pending patent applications, the named applicant(s) were the first to make the inventions claimed in such patents or pending patent applications or that the named applicant(s) were the first to file for patent protection for such inventions.
Further, the issuance, scope, validity, enforceability and commercial value of the Company’s and its current or future licensors’, licensees’ or collaboration partners’ patent rights are highly uncertain. The Company and any potential licensors’ pending and future patent applications may not result in patents being issued that protect the Company’s therapies, in whole or in part, or that effectively prevent others from commercializing competitive technologies and therapies.
Moreover, in some circumstances, the Company may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain such patents, should the Company’s license technology from or to third parties and would be reliant on its licensors, licensees or collaboration partners. If the Company engages with licensors, licensees or collaboration partners and they fail to establish, maintain or protect such patents and other intellectual property rights, such rights may be reduced or eliminated. If such licensors, licensees or collaboration partners were not fully cooperative or disagree with the Company as to the prosecution, maintenance or enforcement of any patent rights, such patent rights could be compromised.
The patent examination process may require the Company or its licensors, licensees or collaboration partners to narrow the scope of the claims of the Company or the Company’s licensors’, licensees’ or collaboration partners’ pending and future patent applications, which may limit the scope of patent protection that may be obtained. The Company cannot guarantee that all of the potentially relevant prior art relating to its patents and patent applications has been found. If such prior art exists, it can invalidate a patent or prevent a patent from issuing from a pending patent application.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and the Company’s patents may be challenged in the courts or patent offices in the UK and abroad. Even if patents do successfully issue and even if such patents cover the Company’s current and future therapeutic candidates, third parties may initiate an opposition, interference, re-examination, post-grant review, inter parties review, nullification or derivation proceedings in court or before patent offices, or similar proceedings challenging the validity, enforceability or scope of such patents, which may result in the patent claims being narrowed or invalidated.
The Company and the Company’s licensors’, licensees’ or collaboration partners’ patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications, and then only to the extent the issued claims cover the technology. In addition, patents and other intellectual property rights also will not protect the Company’s current and any future therapeutic candidates if third parties, including the Company’s competitors, design around the Company’s protected technology and the Company’s current and any future therapeutic candidates without infringing, misappropriating or otherwise violating the Company’s patents or other intellectual property rights. Moreover, some of the Company’s patents and patent applications may in the future be co-owned with third parties. If the Company is unable to obtain an exclusive license to any such third-party co-owners’ interest in such patents or patent applications, such co-owners may be able to license their rights to other third parties, including the Company’s competitors, and the Company’s competitors could market competing therapies and technology. In addition, the Company may need the cooperation of any such co-owners of its patents in order to enforce such patents against third parties, and such cooperation may not be provided. Any of the foregoing could have a material adverse effect on the Company’s competitive position, business, financial conditions, results of operations, and prospects.
Because patent applications are confidential for a period of time after filing, and some remain so until issued, the Company cannot be certain that the Company or its current or future licensors, licensees or collaborators were or will be the first to file any patent application related to a therapeutic candidate. Even where the Company has a valid and enforceable patent, it may not be able to exclude others from practicing the Company’s invention where the other party can show that they used the invention in commerce before the Company’s filing date or the other party benefits from a compulsory license. In addition, the Company may be subject to third-party challenges regarding the Company’s exclusive ownership of the Company’s intellectual property. If a third party were successful in challenging the Company’s exclusive ownership of any of the Company’s intellectual property, the Company may lose its right to use such intellectual property, such third party may be able to license such intellectual property to other third parties, including the Company’s competitors, and the Company’s competitors could market competing therapies and technology. Any of the foregoing could have a material adverse effect on the Company’s competitive position, business, financial conditions, results of operations, and prospects.
As is the case with other biotechnology and pharmaceutical companies, the Company’s success is heavily dependent on intellectual property rights, particularly patents. Obtaining and enforcing patents in the breakthrough neuropsychiatry industry is a technologically and legally complex process, and obtaining and enforcing breakthrough neuropsychiatry patents is costly, time consuming and inherently uncertain. Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of the Company’s and its licensors’ or collaborators’ patent applications and the enforcement or defense of the Company or its licensors’ or collaborators’ issued patents.
Patent Litigation and Intellectual Property
As disclosed under Description of the Business - Intellectual Property, the Company has filed a number of provisional patent applications but even if regular patent applications are filed claiming priority to one or more of the provisional patent applications, there can be no assurance that any or all of these patent applications will issue into a valid patent. Such failure to issue could have a material adverse effect on the Company. In the event that a patent issued to the Company is challenged, any of the Company’s patents may be invalidated. The Company could also become involved in interference or impeachment proceedings in connection with one or more of its patents or patent applications to determine priority of invention.
Patent litigation is widespread in the pharmaceutical industry and the Company cannot predict how this will affect its efforts to form strategic alliances, conduct clinical testing, or manufacture and market any of its prescription drug product candidates that it may successfully develop. If the Company becomes involved in any litigation, interference, impeachment or other administrative proceedings, it will likely incur substantial expenses and the efforts of its technical and management personnel will be significantly diverted. The Company cannot make any assurances that it will have the financial or other resources necessary to enforce or defend a patent infringement or proprietary rights violation action. Moreover, if the Company’s products infringe patents, trademarks or proprietary rights of others, it could, in certain circumstances, become liable for substantial damages, which also could have a material adverse effect on the business of the Company, its financial condition and results of operation. Patent litigation is less likely during development as many jurisdictions contain exemptions from patent infringement for the purpose of obtaining regulatory approval of a product. Where there is any sharing of patent rights either through co-ownership or different licensed “fields of use”, one owner’s actions could lead to the invalidity of the entire patent. If the Company is unable to avoid infringing the patent rights of others, the Company may be required to seek a licence, defend an infringement action or challenge the validity of the patents in court. Such results could have a material adverse effect on the Company. Regardless of the outcome,
patent litigation is costly and time consuming. In some cases, the Company may not have sufficient resources to bring these actions to a successful conclusion, and, even if the Company is successful in these proceedings, it may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on the Company.
Any infringement or misappropriation of the Company’s intellectual property could damage its value and limit its ability to compete. In addition, the Company’s ability to enforce and protect its intellectual property rights may be limited in certain countries outside the U.S., which could make it easier for competitors to capture market position in such countries by utilizing technologies that are similar to those developed or licensed by the Company. Competitors may also harm the Company’s sales by designing products that mirror the capabilities of its products or technology without infringing on its intellectual property rights. If the Company does not obtain sufficient protection for its intellectual property, or if it is unable to effectively enforce its intellectual property rights, its competitiveness could be impaired, which would limit its growth and future revenue. The Company may also find it necessary to bring infringement or other actions against third parties to seek to protect its intellectual property rights. Litigation of this nature, even if successful, is often expensive and time- consuming to prosecute and there can be no assurance that the Company will have the financial or other resources to enforce its rights or be able to enforce its rights or prevent other parties from developing similar technology or designing around its intellectual property.
The Company is not aware of any infringement by it of any person’s or entity’s intellectual property rights. In the event that products sold by the Company are deemed to infringe upon the patents or proprietary rights of others, the Company could be required to modify its products or obtain a licence for the manufacture and/or sale of such products or cease selling such products. In such event, there can be no assurance that the Company would be able to do so in a timely manner, upon acceptable terms and conditions, or at all, and the failure to do any of the foregoing could have a material adverse effect upon the Company’s business. If the Company’s products or proposed products are deemed to infringe or likely to infringe upon the patents or proprietary rights of others, the Company could be subject to injunctive relief and, under certain circumstances, become liable for damages, which could also have a material adverse effect on the Company’s business and its financial condition.
Protection of Intellectual Property
The Company will be able to protect its intellectual property from unauthorized use by third parties only to the extent that the Company’s proprietary technologies, key products and any future products are covered by valid and enforceable intellectual property rights including patents or are effectively maintained as trade secrets and provided the Company has the funds to enforce its rights, if necessary.
To protect the Company’s competitive position, the Company may from time to time need to resort to litigation in order to enforce or defend any patents or other intellectual property rights owned by or licensed to the Company from time to time, or to determine or challenge the scope or validity of patents or other intellectual property rights of third parties. Enforcement of intellectual property rights is difficult, unpredictable and expensive, and many of the Company’s or the Company’s licensors’ or collaboration partners’ adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than the Company or the Company’s licensors or collaboration partners can. Accordingly, despite the Company’s or the Company’s licensors’ or collaboration partners’ efforts, the Company or the Company’s licensors or collaboration partners may not prevent third parties from infringing upon, misappropriating or otherwise violating intellectual property rights. In the event that products sold by the Company own or control, particularly in countries where the laws may not protect
those rights as fully as in the UK, EU, the US and Canada. The Company may fail in enforcing its rights, in which case the Company’s competitors and other third parties may be permitted to use the Company’s therapies without payment to the Company.
In addition, litigation involving the Company’s licensed patents carries the risk that one or more of the Company’s licensed patents will be narrowed, held invalid (in whole or in part, on a claim-by-claim basis) or held unenforceable. Such an adverse court ruling could allow third parties to commercialize the Company’s therapies, and then compete directly with the Company, without payment to the Company.
If the Company were to initiate legal proceedings against a third party to enforce a patent covering one of the Company’s investigational therapies, the defendant could counterclaim that the Company’s patent is invalid or unenforceable. In patent litigation in the UK, EU, the US or Canada, defendant counterclaims alleging invalidity or unenforceability are commonplace. A claim for a validity challenge may be based on failure to meet any of several statutory requirements, for example, lack of novelty, obviousness or non-enablement. A claim for unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the UK Intellectual Property Office, European Patent Office, the USPTO, the CIPO or made a misleading statement, during prosecution. Third parties may also raise challenges to the validity of the Company’s patent claims before administrative bodies in the US or abroad, even outside the context of litigation. Such mechanisms include re-examination, post-grant review, inter partes review, interference proceedings, derivation proceedings, and equivalent proceedings in foreign jurisdictions (i.e., opposition proceedings). Such proceedings could result in the revocation of, cancellation of, or amendment to the Company’s patents in such a way that they no longer cover the Company’s current or any future therapeutic candidates. The outcome following legal assertions of invalidity and unenforceability during patent litigation or other proceedings is unpredictable. With respect to the validity question, for example, the Company cannot be certain that there is no invalidating prior art, of which the Company and the patent examiner were unaware during prosecution. If a defendant or third party were to prevail on a legal assertion of invalidity or unenforceability, the Company would lose at least part, and perhaps all, of the patent protection on the Company’s current or one or more of any future therapeutic candidates. Such a loss of patent protection could have a material adverse impact on the Company’s business financial condition, results of operations, and prospects. Further, litigation could result in substantial costs and diversion of management resources, regardless of the outcome, and this could harm the Company’s business and financial results.
Third-Party Licences
A substantial number of patents have already been issued to other biotechnology and pharmaceutical companies. To the extent that valid third-party patent rights cover the Company’s products or services, the Company or its strategic collaborators would be required to seek licences from the holders of these patents in order to manufacture, use or sell these products and services and payments under them would reduce the Company’s profits from these products and services. The Company is currently unable to predict the extent to which it may wish or be required to acquire rights under such patents, the availability and cost of acquiring such rights and whether a licence to such patents will be available on acceptable terms or at all. There may be patents in the U.S. or in foreign countries or patents issued in the future that are unavailable to licence on acceptable terms. The Company’s inability to obtain such licences may hinder or eliminate its ability to manufacture and market its products.
Further, if the Company obtains third-party licences but fails to pay annual maintenance fees, development and sales milestones, or it is determined that the Company does not use commercially
reasonable efforts to commercialize licensed products, the Company could lose its licences which could have a material adverse effect on its business and financial condition.
Periodic maintenance and annuity fees on any issued patent are due to be paid to the UK Intellectual Property Office, the European Patent Office, the USPTO, the CIPO and foreign patent agencies in several stages over the lifetime of the patent. The European Patent Office, the USPTO, the CIPO and various foreign governmental patent agencies also require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In certain circumstances, the Company may rely on collaboration partners to pay these fees due to US and comparable foreign patent agencies and take the necessary action to comply with such requirements with respect to the Company’s intellectual property. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If the Company, its licensors or collaboration partners fail to maintain the patents and patent applications covering the Company’s investigational therapies, third parties, including its competitors might be able to enter the market with similar or identical therapies or technologies, which would have a material adverse effect on the Company’s business, financial condition, results of operations, and prospects.
FINANCIAL AND ACCOUNTING RISKS
Substantial Number of Authorized but Unissued Common Shares
The Company has an unlimited number of Common Shares that may be issued by the Board without further action or approval of the Shareholders. While the Board will be required to fulfill its fiduciary obligations in connection with the issuance of such Common Shares, the Common Shares may be issued in transactions with which not all of the shareholders of the Company agree, and the issuance of such Common Shares will cause dilution to the ownership interests of the shareholders of the Company.
Dilution
The Company may issue additional Common Shares in subsequent offerings (including through the sale of securities convertible into or exchangeable for Common Shares) and on the exercise of stock options or other securities exercisable for Common Shares. The Company cannot predict the size of future issuances of Common Shares or the effect that future issuances and sales of Common Shares will have on the market price of the Common Shares. Issuances of a substantial number of additional Common Shares, or the perception that such issuances could occur, may adversely affect prevailing market prices for the Common Shares. With any additional issuance of Common Shares, investors will suffer dilution to their voting power and the Company may experience dilution in its earnings per share, including as a result of issuances under the Equity Incentive Plan, convertible or exchangeable securities, or other corporate arrangements, including the Rights Plan (as defined herein).
Negative Cash Flow from Operating Activities
The Company has had negative cash flow from operating activities since inception. Drug development involves long lead times, is very expensive and involves many variables of uncertainty. As such, significant capital investment will be required to achieve the Company’s existing plans. The Company’s
net losses have had and will continue to have an adverse effect on, among other things, shareholder equity, total assets and working capital. The Company expects that losses may fluctuate from quarter to quarter and year to year, and that such fluctuations may be substantial based on the stage of development of its principal programs. The Company cannot predict when it will become profitable, if at all. Accordingly, the Company may be required to obtain additional financing in order to meet its future cash commitments.
Additional Capital Requirements
As a research and development company, the Company expects to spend substantial funds to continue the research, development and testing of its prescription drug product candidates and to prepare to commercialize products subject to applicable regulatory approval. Substantial additional financing may be required if the Company is to be successful in continuing to develop its business and its products. No assurances can be given that the Company will be able to raise the additional capital that it may require for its anticipated future development. Any additional equity financing may be dilutive to investors and debt financing, if available, may involve restrictions on financing and operating activities. There is no assurance that additional financing will be available on terms acceptable to the Company, if at all. If the Company is unable to obtain additional financing as needed, it may be required to reduce the scope of its operations or anticipated expansion. The Company’s ability to successfully raise additional capital and maintain liquidity may by impaired by factors outside of its control, such as a shift in consumer attitudes towards certain therapeutic methods or a downturn in the economy.
Heightened regulatory scrutiny could have a negative impact on the Company’s ability to raise capital. The Company’s business activities rely on developing laws and regulations in multiple jurisdictions. It is impossible to determine the extent of the impact of any new laws, regulations or initiatives that may be proposed, or whether any proposals will become law. The regulatory uncertainty surrounding the Company’s current or any future products may adversely affect the Company’s business and operations, including without limitation, the Company’s ability to raise additional capital.
In addition, the Company may encounter unforeseen expenses, difficulties, complications, delays and other known or unknown factors in achieving its business objectives. The Company will eventually need to transition from a company with a development focus to a company capable of supporting commercial activities. The Company may not be successful in such a transition.
Lack of Significant Product Revenue
To date, the Company has generated little product revenue and cannot predict when and if it will generate significant product revenue. The Company’s ability to generate significant product revenue and ultimately become profitable depends upon its ability, alone or with partners, to successfully develop its prescription drug product candidates, obtain regulatory approval and commercialize products, including any of its current prescription drug product candidates or other prescription drug product candidates that it may develop, in-license or acquire in the future. The Company does not anticipate generating revenue from the sale of products for the foreseeable future. The Company expects its research and development expenses
to increase in connection with its ongoing activities, particularly as it advances its prescription drug product candidates through clinical trials.
Estimates or Judgments Relating to Critical Accounting Policies
The preparation of financial statements in conformity with the International Financial Reporting Standards requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. The Company bases its estimates on historical experience and on various other assumptions that it believes to be reasonable under the circumstances, as provided in the notes to the financial statements of the Company, the results of which form the basis for making judgments about the carrying values of assets, liabilities, equity, revenue and expenses that are not readily apparent from other sources. The Company’s operating results may be adversely affected if the assumptions change or if actual circumstances differ from those in the assumptions, which could cause its operating results to fall below the expectations of securities analysts and investors, resulting in a decline in the share price of the Company. Significant assumptions and estimates used in preparing the financial statements include those related to income tax credits receivable, share based payments, impairment of non-financial assets, fair value of biological assets, as well as cost recognition.
Inadequate Internal Controls
If the Company fails to maintain an effective system of internal controls, the Company might not be able to report its financial results accurately or prevent misstatement; and in that case, the Company’s shareholders could lose confidence in its financial reporting, which would harm its business and could negatively impact the value of its shares. While the Company believes that it has sufficient personnel and review procedures to allow it to maintain an effective system of internal controls, there can be no assurance that the Company will always successfully detect misstatements or implement necessary improvements in a timely fashion.
RISKS RELATED TO THE COMMON SHARES
Market for the Common Shares
There can be no assurance that an active trading market for the Common Shares will develop or, if developed, that any market will be sustained. The Company cannot predict the prices at which the Common Shares will trade. Fluctuations in the market price of the Common Shares could cause an investor to lose all or part of its investment in Common Shares. Factors that could cause fluctuations in the trading price of the Common Shares include: (i) announcements of new offerings, products, services or technologies; commercial relationships, acquisitions or other events by the Company or its competitors; (ii) price and volume fluctuations in the overall stock market from time to time; (iii) significant volatility in the market price and trading volume of companies commercializing serotonergic agonist pharmaceuticals; (iv) fluctuations in the trading volume of the Common Shares or the size of the Company’s public float; (v) actual or anticipated changes or fluctuations in the Company’s results of operations; (vi) whether the Company’s results of operations meet the expectations of securities analysts or investors; (vii) actual or anticipated changes in the expectations of investors or securities analysts; (viii) litigation involving the Company, its industry, or both; (ix) regulatory developments; (x) general economic conditions and trends; (xi) major catastrophic events; (xii) escrow releases, sales of large blocks of the Common Shares; (xiii) departures of key employees or members of management; or (xiv) an adverse impact on the Company from any of the other risks cited herein.
Significant Sales of the Common Shares
Although Common Shares held by existing shareholders of the Company will be freely tradable under applicable securities legislation, the Common Shares held by the Company’s directors, executive officers, Control persons and certain other security holders may be subject to contractual lock-up restrictions and may also be subject to escrow restrictions pursuant to the policies of Cboe Canada. Sales of a substantial number of the Common Shares in the public market after the expiry of lock-up or escrow restrictions, or the perception that these sales could occur, could adversely affect the market price of the Common Shares and may make it more difficult for investors to sell Common Shares at a favourable time and price.
Volatile Market Price for the Common Shares
The securities market in Canada has experienced a high level of price and volume volatility, and the market prices of securities of many companies have experienced wide fluctuations in price which have not necessarily been related to the operating performance, underlying asset values or prospects of such companies. There can be no assurance that continual fluctuations in price will not occur. It may be anticipated that any market for the Common Shares will be subject to market trends generally, notwithstanding any potential success of the Company. The value of the Common Shares distributed hereunder will be affected by such volatility.
The volatility of the Common Shares may affect the ability of holders to sell the Common Shares at an advantageous price or at all. Market price fluctuations in the Common Shares may be adversely affected by a variety of factors relating to the Company’s business, including fluctuations in the Company’s operating and financial results, such results failing to meet the expectations of securities analysts or investors and downward revisions in securities analysis’ estimates in connection therewith, sales of additional Common Shares, governmental regulatory action, adverse change in general market conditions or economic trends, acquisitions, dispositions or other material public announcements by the Company or its competitors, along with a variety of additional factors, including, without limitation, those set forth under the heading “Cautionary Note Regarding Forward-Looking Information”. In addition, the market price for securities on stock markets, including Cboe Canada is subject to significant price and trading fluctuations. These fluctuations have resulted in volatility in the market prices of securities that often has been unrelated or disproportionate to changes in operating performance. These broad market fluctuations may materially adversely affect the market price of the Company.
Additionally, the value of the Common Shares is subject to market value fluctuations based upon factors that influence the Company’s operations, such as legislative or regulatory developments, competition, technological change and changes in interest rates or foreign exchange rates. There can be no assurance that the market price of the Common Shares will not experience significant fluctuations in the future, including fluctuations that are unrelated to the Company’s performance.
Tax Issues
There may be income tax consequences in relation to the Common Shares, which will vary according to circumstances. Independent advice from tax and legal advisers should be obtained.
No Dividends
The Company’s current policy is, and will be, to retain earnings to finance the development and enhancement of its products and to otherwise reinvest in the Company. Therefore, the Company does not anticipate paying cash dividends on the Common Shares in the foreseeable future. The Company’s
dividend policy will be reviewed from time to time by the Board in the context of its earnings, financial condition and other relevant factors. Until the time that the Company does pay dividends, which it might never do, its shareholders will not be able to receive a return on their Common Shares unless they sell them.
DIVIDEND AND DISTRIBUTIONS
The Company does not currently intend to declare any dividends payable to the holders of the Common Shares. The Company has no restrictions on paying dividends, but if the Company generates earnings in the foreseeable future, it expects that they will be retained to finance growth. The Board will determine if and when dividends should be declared and paid in the future based upon the Company’s financial position at the relevant time.
DESCRIPTION OF CAPITAL STRUCTURE
As of the date of this AIF, the authorized share capital of the Company consists of an unlimited number of Common Shares of which 61,984,078 are issued and outstanding, and an unlimited number of preferred shares, issuable in series, none of which are issued and outstanding.
In addition, the Company had agreed to issue Common Shares in connection with the Adelia Transaction. The Common Shares were issuable upon exchange of Class B Shares in the capital of Helus US on the basis of 0.26316 Common Shares for 1 Class B Share, subject to customary adjustments. The Adelia Shareholders were also entitled to Class B Shares upon the occurrence of certain milestones. No Class B Shares were exchangeable prior to the first anniversary of closing of the Adelia Transaction, and not more than: (i) 33 1/3% of the Class B Shares were exchangeable prior to the second anniversary of the Adelia Transaction; and (ii) 66 2/3% of the Class B Shares were exchangeable prior to the third anniversary of the Adelia Transaction. Helus US has issued 1,591,625.3 Class B Shares, of which 1,555,540.6 have been exchanged into Common Shares, and the remaining 36,084.7 Class B Shares were cancelled, effective August 20, 2025. For further information see “General Development of the Business – Three Year History” and “Prior Sales – Exchangeable Securities”.
Holders of Common Shares are entitled to one vote for each Common Share held at all meetings of shareholders of the Company, to receive dividends if, as and when declared by the Board, and to participate ratably in any distribution of property or assets upon the liquidation, winding-up or other dissolution of the Company. The Common Shares carry no pre-emptive rights, conversion or exchange rights, or redemption, retraction, repurchase, sinking fund or purchase fund provisions. There are no provisions requiring a holder of Common Shares to contribute additional capital, and no restrictions on the issuance of additional securities by the Company. There are no restrictions on the repurchase or redemption of Common Shares by the Company except to the extent that any such repurchase or redemption would render the Company insolvent.
The aim of the Equity Incentive Plan is to attract and retain employees, directors and consultants, and to ensure that interests of key persons are aligned with the success of the Company and its affiliates. The maximum number of options to purchase Common Shares reserved for issuance under the Equity Incentive Plan pursuant to options not intended as incentive stock options shall be 20% of the issued and outstanding Common Shares from time to time, on a non-diluted basis. The maximum number of Common Shares reserved for issuance under the Equity Incentive Plan pursuant to incentive stock options is 3,998,381. For the avoidance of doubt, long-term incentive options are excluded from the Equity Incentive Plan maximum. Common Shares in respect of Options that have been exercised, cancelled,
surrendered, or terminated or that expire without being exercised shall again be available for issuance under the Equity Incentive Plan.
The Board has adopted a shareholder rights plan (the “Rights Plan”) to ensure, to the extent possible, that all shareholders of the Company and the Board have adequate time to consider and evaluate any unsolicited take-over bid for the Company, provide the Board with adequate time to evaluate any such take-over bid and explore and develop value-enhancing alternatives to any such take-over bid, encourage the fair treatment of the Company’s shareholders in connection with any such take-over bid, and generally assist the Board in enhancing shareholder value. The Rights Plan was approved by the Company’s shareholders on, and took effect as of, August 16, 2021. An amended and restated Rights Plan was approved by shareholders at the Company’s annual and special meeting of shareholders held on August 27, 2024.
MARKET FOR SECURITIES
Trading Price and Volume
The Common Shares were listed for trading on Cboe Canada and on NYSE American under the trading symbol “CYBN”. On January 5, 2026, the Company transferred its U.S. stock exchange listing from NYSE American to Nasdaq under the ticker symbol “HELP” see “Corporate Structure – Name, Address and Incorporation”. The Company continues to be listed on Cboe Canada under the same “HELP” ticker symbol.
The following tables set forth, for the periods indicated, the reported monthly high and low prices per Common Share and the total monthly trading volumes on Cboe Canada, NYSE American, and Nasdaq, as applicable, for the most recently completed financial year end.
| | | | | | | | | | | | | | | | | | | | |
Cboe Canada Price Range |
Month(1) |
| High (CA$) |
| Low (CA$) |
| Volume |
|
| |
| |
| |
| April 2025 | | 11.12 | | 6.95 | | 223,736 |
| May 2025 | | 11.74 | | 8.40 | | 217,266 |
| June 2025 | | 12.56 | | 9.85 | | 189,178 |
| July 2025 | | 12.18 | | 9.98 | | 261,253 |
| August 2025 | | 10.90 | | 8.80 | | 229,553 |
| September 2025 | | 10.33 | | 8.00 | | 451,016 |
| October 2025 | | 10.77 | | 8.05 | | 469,093 |
| November 2025 | | 10.41 | | 7.85 | | 471,080 |
| December 2025 | | 11.99 | | 7.70 | | 465,560 |
| January 2026 | | 12.12 | | 8.89 | | 315,582 |
| February 2026 |
| 10.75 | | 7.90 | | 316,499 |
| March 2026 | | 11.61 | | 5.90 | | 475,838 |
Note:
(1) Source: Cboe Canada trading data as of the date of this AIF. All figures related to securities are represented on a post-Consolidation basis.
As the close of business on June 26, 2026, being the last day on which the Common Shares traded prior to the date of this AIF, the price of the Common Shares as quoted by Cboe Canada was CA$9.17 per Common Share.
| | | | | | | | | | | | | | | | | | | | |
NYSE American / Nasdaq Price Range |
Month(1) |
| High (US$)(1) |
| Low (US$)(1) |
| Volume(1) |
|
| |
| |
| |
April 2025(2) | | 8.09 | | 4.81 | | 6,401,818 |
May 2025(2) | | 8.50 | | 6.05 | | 6,742,091 |
June 2025(2) | | 9.20 | | 7.11 | | 5,711,894 |
July 2025(2) | | 9.83 | | 7.26 | | 11,021,801 |
August 2025(2) | | 7.88 | | 6.36 | | 7,812,206 |
September 2025(2) | | 7.44 | | 5.72 | | 15,583,822 |
October 2025(2) | | 8.00 | | 5.71 | | 22,869,736 |
November 2025(2) | | 7.44 | | 5.52 | | 14,108,797 |
December 2025(5) | | 8.71 | | 5.51 | | 19,897,907 |
January (1-2) 2026(3)(5) | | 8.28 | | 6.62 | | 8,976,315 |
January (5-31) 2026(4)(5) | | 8.97 | | 6.52 | | 8,258,614 |
February 2026(5) |
| 7.83 | | 5.76 | | 12,148,771 |
March 2026(5) | | 8.55 | | 4.29 | | 27,110,747 |
Notes:
(1) All figures related to securities are represented on a post-Consolidation basis.
(2) Common Shares listed for trading on the NYSE American. Source: NYSE American as of the date of this AIF.
(3) Represents trading on the facilities of the NYSE American for the period from January 1, 2026 to January 2, 2026. At market close on January 2, 2026, trading was halted on the NYSE American.
(4) Represents trading on the facilities of Nasdaq for the period from January 5, 2026 to January 31, 2026. At market open on January 5, 2026, trading commenced on Nasdaq.
(5) Source: Nasdaq as of the date of this AIF.
As the close of business on June 26, 2026, being the last day on which the Common Shares traded prior to the date of this AIF, the price of the Common Shares as quoted by Nasdaq was $6.48 per Common Share.
Prior Sales
The following tables summarize details of the following securities that are not listed or quoted on a marketplace issued by the Company during the most recently completed financial year end:
| | | | | | | | | | | |
| Date Granted | Type of Security Issued | Number of Securities Issued | Issuance / Exercise Price (CA$) |
| June 30, 2025 | Convertible Debentures | $50,000 | See note (1) |
August 15, 2025 (2) | Options | 53,800 | $10.00 |
| August 15, 2025 | Options | 80,000 | $11.00 |
| August 29, 2025 | Options | 15,000 | $11.00 |
| October 1, 2025 | Options | 200,000 | $8.39 |
| October 1, 2025 | Restricted Share Units | 600,000 | N/A |
October 31, 2025(3) | Warrants | 9,049,138 | $11.33 |
October 31, 2025(4) | Pre-Funded Warrants | 4,605,500 | $0.000014 |
| November 3, 2025 | Restricted Share Units | 3,564,440 | N/A |
November 14, 2025(5) | Options | 48,240 | $8.39 |
| December 31, 2025 | Options | 13,430 | $11.65 |
February 10, 2026(6) | Restricted Share Units | 975,000 | N/A |
February 10, 2026(7) | Performance Share Units | 325,000 | N/A |
| February 24, 2026 | Restricted Share Units | 25,000 | N/A |
March 10, 2026(8) | Restricted Share Units | 35,000 | N/A |
Notes:
(1)Convertible Debentures issued in connection with the Convertible Debenture Private Placement. The principal amount of, and accrued and unpaid interest, if any, on the Convertible Debentures is convertible into Common Shares at the Conversion Price (as defined in the High Trail Securities Purchase Agreement).
(2)On October 31, 2025, and January 29, 2026, 4,375 and 625 options, respectively, were terminated as a result of the optionee no longer being eligible under the Equity Incentive Plan.
(3)Common Share purchase warrants issued in connection with the Registered Direct Offering.
(4)Pre-funded Common Share purchase warrants issued in connection with the Registered Direct Offering.
(5)On December 19, 2025, 4,000 options were terminated as a result of the optionee no longer being eligible under the Equity Incentive Plan.
(6)On April 19, 2026, 975,000 restricted share units were cancelled as a result of the holder no longer being eligible under the terms of a restricted share unit agreement.
(7)On April 19, 2026, 325,000 performance share units were cancelled as a result of the holder no longer being eligible under the Equity Incentive Plan.
(8)On May 13, 2026, 35,000 restricted share units were cancelled as a result of the holder no longer being eligible under the terms of a restricted share unit agreement.
Exchangeable Securities
During the year ended March 31, 2026, no additional Class B shares were issued. As of March 31, 2026, all Milestones were achieved, and all eligible Class B shares were issued.
DIRECTORS AND EXECUTIVE OFFICERS
The following table lists the names and municipalities of residence of the directors and officers of the Company, their positions and offices to be held with the Company, their principal occupations during the past five years and the number of securities of the Company that are beneficially owned, directly or indirectly, or over which control or direction will be exercised by each such director or officer. Each of the directors is elected to hold office until the next annual meeting of the shareholders of the Company or until a successor is duly elected or appointed.
| | | | | | | | | | | |
| Name, Municipality of Residence and Position Held | Principal Occupation for the Past Five Years | Appointed to Position Held | Number and Percentage of Securities Beneficially Owned or Controlled |
Greg Cavers, Toronto, Ontario, Canada
Chief Financial Officer | Chief Financial Officer, Helus Pharma
| November 2020 | 18,987(7) (0.03%) |
Gabriel Fahel, Ottawa, Ontario, Canada
Chief Legal Officer | Chief Legal Officer, Helus Pharma
| November 2020 | 38,451(8) (0.06%) |
Aaron Bartlone, Milton, Georgia, United States
Chief Operating Officer | Chief Operating Officer, Helus Pharma
| July 2023 | 34,321(9) (0.06%) |
Paul Glavine, United Arab Emirates
Director and Chief Growth Officer | Chief Growth Officer, Helus Pharma
Former Chief Operating Officer and Chief Executive Officer, Helus Pharma
| November 2020 March 2021 | 299,674(10) (0.48%) |
Eric So(4), United Arab Emirates
Director, President and Interim CEO | President and Interim CEO, Helus Pharma
| November 2020 | 311,115(11) (0.50%) |
Theresa Firestone(1)(2)(3)(5), Toronto, Ontario, Canada
Director | Independent Board Director
| August 2021 | 26,059 (0.04%) |
Grant Froese(1)(2), Toronto, Ontario, Canada
Director | Principal, Greywolf Management Services Inc.
Director and Chief Executive Officer, Harvest One Cannabis Inc. | November 2020 | 30,263(12) (0.05%) |
| | | | | | | | | | | |
| Name, Municipality of Residence and Position Held | Principal Occupation for the Past Five Years | Appointed to Position Held | Number and Percentage of Securities Beneficially Owned or Controlled |
Eric Hoskins(1)(3)(4)(6), Toronto, Ontario, Canada
Director | Partner, Maverix Private Equity | November 2020 | 27,632(13) (0.04%) |
Mark Lawson(1)(2)(3)(4), Toronto, Ontario, Canada
Director | Chief Growth Officer, Invert Inc.
Managing Partner, Clermont Capital Partners | November 2020 | 28,026 (0.05%) |
George Tziras, London, United Kingdom
Chief Business Officer | Chief Business Officer, Helus Pharma
Chief Executive Officer and Chief Business Officer, Small Pharma Ltd.
Executive Director, Goldman Sachs International | October 2023 | 36,270(14) (0.06%) |
Freda Lewis-Hall(6), Naples, Florida, United States
Director | Retired | February 2026 | Nil(15) |
Amir Inamdar, Tiptree(16), Essex, United Kingdom
Chief Medical Officer | Chief Medical Officer, Helus Pharma
| October 2021 | 20,156(16) (0.03%) |
Notes:
(1) Member of the Audit Committee.
(2) Member of the Compensation Committee.
(3) Member of Governance and Nominating Committee.
(4) Member of the Transactions Committee.
(5) Lead independent director.
(6) Member of the Scientific Advisory Committee.
(7) Excludes 83,334 RSUs to acquire 83,334 Common Shares.
(8) Excludes 83,334 RSUs to acquire 83,334 Common Shares.
(9) Excludes 83,334 RSUs to acquire 83,334 Common Shares.
(10) Excludes 105,263 Warrants to acquire 105,263 Common Shares and 1,907,439 RSUs to acquire 1,907,439 Common Shares.
(11) Excludes 105,264 Warrants to acquire 105,264 Common Shares and 1,907,439 RSUs to acquire 1,907,439 Common Shares.
(12) Excludes 19,737 Warrants to acquire 19,737 Common Shares.
(13) Excludes 30,263 Warrants to acquire 30,263 Common Shares.
(14) Excludes 39,474 Options to acquire 39,474 Common Shares, and 20,834 RSUs to acquire 20,834 Common Shares.
(15) Excludes 25,000 Options to acquire 25,000 Common Shares and 25,000 RSUs to acquire 25,000 Common Shares.
(16) Excludes 56,779 Options to acquire 56,779 Common Shares and 83,334 RSUs to acquire 83,334 Common Shares.
As of the date of this AIF, all promoters, directors, officers and insiders, as a group, beneficially own, directly or indirectly, an aggregate of 870,594 Common Shares on a non-diluted basis, representing 1.41% of the Company’s capitalization on a non-diluted basis.
Board of Directors & Management
Greg Cavers, Chief Financial Officer, Age 56
Greg Cavers has over 20 years’ experience specializing in transforming and revitalizing corporate finance departments. Mr. Cavers has experience in service operations in varying stages of growth; leading business unit start-ups, restructuring, system implementations and merger integrations while increasing profitability, minimizing risk and dedicated to meeting financial reporting, IFRS; as well as regulatory reporting OSFI, MFDA requirements.
Gabriel Fahel, Chief Legal Officer and Corporate Secretary, Age 51
Gabriel (Gabe) Fahel is a seasoned legal executive and a central architect of the corporate evolution and growth of Helus Pharma. With over 25 years of experience navigating complex corporate-commercial matters, government relations, and international public structures, Gabe has played a pivotal role in bridging the gap between innovative small-molecule pharmaceuticals and global capital markets. Since the company’s inception, he has shepherded Helus Pharma through its most transformative milestones, including leading the legal execution for more than $500 million in total capital raised and overseeing the Company’s public listings on both the NYSE and Nasdaq.
Aaron Bartlone, Chief Operation Officer, Age 59
Aaron Bartlone is an accomplished and highly regarded biopharmaceutical executive with an impressive track record of driving disruptive therapies to worldwide markets. Prior to joining the Company, Aaron served UCB for 12 years as Senior Vice President of Quality Assurance, Patient Safety and Enterprise Risk Management, where he delivered functional results, clinical pipeline progression and regulatory marketing approvals for products in neurology, gastroenterology and immunology. Aaron also served as President of the US Commercial Operations where he delivered double-digit growth on a +US$2 billion P&L while concurrently restructuring the business units and supporting compliance infrastructure. Before UCB, Aaron spent 14 years with Eli Lilly & Company across a myriad of US and global roles spanning Quality Assurance, Regulatory Affairs and CMC Product Development, where he drove successful neurology, oncology, diabetes, and cardiovascular drug candidates and combination products through the clinical phases to global markets. Aaron holds a bachelor’s degree in Chemistry & Mathematics from Youngstown State University and a master’s degree in Analytical Chemistry from the University of Notre Dame.
Paul Glavine, Co-founder, Chief Growth Officer, and Director, Age 37
Paul Glavine is a biotechnology entrepreneur and investor leading growth at Helus Pharma, a clinical-stage biopharmaceutical company developing novel serotonergic agonists for mental healthcare. Under his leadership, Helus Pharma secured FDA Breakthrough Therapy Designation for HLP003 and advanced HLP004, a generalized anxiety disorder candidate, from preclinical studies into human trials. A serial entrepreneur, Mr. Glavine has raised over $500 million in capital and led mergers and acquisitions that have expanded operating businesses and therapeutic pipelines across his portfolio. He serves as Chairman
of LongPoint ETFs. He is a Milken Institute Young Leader and was named to Catalyst Health’s Top 40 Under 40 in Life Sciences.
Eric So, Co-founder, President, Interim CEO and Director, Age 50
Eric So is a veteran founder and operator of various public and private companies over the last 20 years and has led corporate strategy, development and finance at all stages of the business life cycle from start-up to high growth and large multi-national. A trusted advisor he began his career practicing in the areas of corporate commercial, securities, finance and mergers and acquisitions at a leading international law firm. He has focused on sectors which have profound global impact in critical areas such as mental health and addiction.
George Tziras, Chief Business Officer, Age 45
Mr. Tziras has over 15 years of experience in investment banking and international capital markets having worked at a number of global financial institutions including Goldman Sachs, Credit Suisse, Nomura, Lehman Brothers and CIBC. Mr. Tziras has worked on a broad range of transactions including debt and equity financings; mergers, disposals and acquisitions; private equity buyouts and debt restructurings. He has also worked across a number of industries, including healthcare. Mr. Tziras holds a BA degree from the University of Oxford and a MA degree from Johns Hopkins. Mr. Tziras joined Small Pharma in 2021 as Chief Business Officer and on July 20, 2022, transitioned to Chief Executive Officer of Small Pharma until it was acquired by the Company on October 23, 2023. Mr. Tziras has been a director of Small Pharma Ltd (now Cybin UK Ltd. T/A Helus) since 2015. Mr Tziras joined Helus Pharma on October 24, 2023.
Amir Inamdar, Chief Medical Officer, Age 52
Amir Inamdar, MBBS, DNB (Psych), FFPM, is a qualified psychiatrist and pharmaceutical physician with over 25 years of experience in clinical research and drug development. Dr. Inamdar has successfully progressed numerous candidate drugs from preclinical development and early-phase clinical trials to proof-of-concept studies and through to successful marketing authorization. He has extensive experience leading global, cross-functional teams in the development of novel medications across a range of psychiatric indications, including treatment resistant depression, narcolepsy, anxiety, schizophrenia, bipolar disorder and substance use disorders.
Theresa Firestone, Director, Age 70
Ms. Theresa Firestone is a senior healthcare executive with over 35 years’ experience in pharmaceuticals, health & wellness and government and has extensive P&L, strategy development and operations expertise. Ms. Firestone has held executive leadership positions in Canada, Europe and Asia and led teams in 15 different countries. Prior to retirement in 2021, she was Senior Vice President, Health and Wellness at Shoppers Drug Mart (SDM), Canada’s largest retail pharmacy chain. Prior to Shoppers, Ms. Firestone was Regional President of Emerging Markets Asia with Pfizer Inc (Shanghai and HK). She was also General Manager of the Established Products Business Unit, Pfizer Canada, Country Manager, Pfizer Austria, VP Sales and VP of Government Affairs with Pfizer Canada. She currently sits on a number of Boards, including Apotex, Prollenium Medical Technologies, adMare BioInnovations and Blue Charm Adherence.
Grant Froese, Director, Age 64
Grant Froese completed a 38-year career with Canadian retail giant Loblaw Companies Limited where he last served as Chief Operating Officer until his retirement. During his career at Loblaw, he led operations, merchandising and had oversight of supply chain, ecommerce, and marketing functions. After retirement Grant was the CEO of Harvest One / Delivra Health Brands until 2020. Currently, Grant is the principal consultant at Grey Wolf Management Services Inc. and sits on the board of several companies.
Eric Hoskins, Director, Age 65
Dr. Eric Hoskins is a Partner at Maverix Private Equity. He is the former Ontario Health Minister (2014-2018) responsible for one of the largest health care systems in North America. He is a former elected Member of Ontario Provincial Parliament holding Cabinet positions in Health, Economic Development and Trade, Children and Youth Services, and Immigration. Dr. Hoskins is a physician and public health specialist with more than thirty years’ experience in health care and public policy.
Mark Lawson, Director, Age 53
Mark Lawson is a private equity and investment banking executive with over 20 years of experience in Canada, the United States, and in the emerging markets. He is currently the Head of Carbon Acquisition for Invert, a company that funds global carbon reduction and removal projects. From 2009 to 2023 Mr. Lawson was the Managing Partner of Clermont Capital Partners, a Toronto based merchant bank and advisory firm focused on the technology and healthcare sectors. From 2004 to 2008 he was an investment banker with Morgan Stanley in New York, where he was involved in the execution of over $6 billion worth of mergers and acquisitions, $8 billion worth of debt offerings and $500 million of equity financings in the healthcare, technology, and telecom sectors. Mr. Lawson is also currently a director of various publicly traded companies in North America. Mr. Lawson received his Bachelor of Arts in Statistical Sciences from The University of Western Ontario, Canada and his MBA from The Richard Ivey School of Business, University of Western Ontario, Canada. Mr. Lawson is a member of the Economic Club of New York and is a Director of the Hugh and Ilene Lawson Charitable Organization.
Dr. Freda Lewis-Hall, Director, Age 71
Dr. Freda Lewis-Hall is a pioneering physician and biopharmaceutical executive with more than 40 years of experience spanning clinical care, research, academia, corporate leadership, and public health advocacy. She began her medical career as a practicing psychiatrist, focusing on the impact of mental illness on families and communities. She later held senior leadership roles across the biopharmaceutical industry, including serving for more than a decade on Pfizer’s Executive Leadership Team as Executive Vice President and Chief Medical Officer. In that role, she led a global medical organization operating in more than 125 countries and supporting a broad portfolio of medicines and vaccines. She subsequently served as Chief Patient Officer, advancing patient engagement, inclusion, and health equity initiatives across the enterprise. During her tenure, she also helped lead the spinout of SpringWorks Therapeutics and served on its Board of Directors, guiding the company through regulatory approvals and its evolution into a commercial-stage organization prior to its acquisition by Merck KGaA for approximately $3.4 billion. Dr. Lewis-Hall has also held senior leadership positions at Vertex Pharmaceuticals, Bristol Myers Squibb, Pharmacia Corporation, and Eli Lilly and Company. Dr. Lewis-Hall is a Distinguished Fellow of the American Psychiatric Association and previously served as Vice Chairperson and Associate Professor in the Department of Psychiatry at Howard University College of Medicine. She has also advised the National Institute of Mental Health.
CEASE TRADE ORDERS, BANKRUPTCIES, PENALTIES OR SANCTIONS
Except as disclosed below, no director or executive officer of the Company is, as at the date of this AIF, or has been within the last ten years, a director, chief executive officer or chief financial officer of any company (including the Company) that:
(a) was subject to a cease trade order, an order similar to a cease trade order, or an order that denied the relevant company access to any exemption under securities legislation, and which in all cases was in effect for a period of more than 30 consecutive days (an “Order”), which Order was issued while the director or executive officer was acting in the capacity as director, chief executive officer or chief financial officer of such company; or
(b) was subject to an Order that was issued after the director or executive officer ceased to be a director, chief executive officer or chief financial officer and which resulted from an event that occurred while that person was acting in the capacity as director, chief executive officer or chief financial officer of such company.
To the knowledge of the Company, no director or executive officer of the Company or any shareholder holding a sufficient number of Common Shares to affect materially the control of the Company:
(a) is, as at the date of this AIF, or has been within the last ten years, a director or executive officer of any company (including the Company) that, while that person was acting in that capacity, or within a year of that person ceasing to act in that capacity, became bankrupt, made a proposal under any legislation relating to bankruptcy or insolvency or was subject to or instituted any proceedings, arrangement or compromise with creditors or had a receiver, receiver manager or trustee appointed to hold its assets;
(b) has, within the last ten years, become bankrupt, made a proposal under any legislation relating to bankruptcy or insolvency or become subject to or instituted any proceedings, arrangement or compromise with creditors or had a receiver, receiver manager or trustee appointed to hold his assets;
(c) has been subject to any penalties or sanctions imposed by a court relating to securities legislation or by a securities regulatory authority or has entered into a settlement agreement with a securities regulatory authority; or
(d) has been subject to any penalties or sanctions imposed by a court or regulatory body that would likely be considered important to a reasonable investor in making an investment decision regarding the Company.
Greg Cavers was the interim Chief Financial Officer of LottoGopher Holdings Inc. (“LottoGopher”), a CSE-listed company, until January 2020. Preceding his position, LottoGopher had been subject to a cease trade order on December 5, 2018 for failing to file interim financial report, management’s discussion and analysis and certification of the filings pursuant to NI 52-109.
The foregoing information, not being within the knowledge of the Company, has been furnished by the respective directors and executive officers.
CONFLICTS OF INTEREST
To the best of the Company’s knowledge, other than as disclosed herein, there are no known existing or potential material conflicts of interest between the Company and any directors or officers of the Company, except that certain of the directors and officers serve as directors, officers, promoters and members of management of other public companies and therefore it is possible that a conflict may arise between their duties as a director or officer of the Company and their duties as a director, officer, promoter or member of management of such other companies.
The directors and officers of the Company are aware of the existence of laws governing accountability of directors and officers for corporate opportunity and requiring disclosures by directors of conflicts of interest and the Company will rely upon such laws in respect of any directors and officers’ conflicts of interest or in respect of any breaches of duty by any of its directors or officers. All such conflicts will be disclosed by such directors or officers in accordance with the OBCA and they will govern themselves in respect thereof to the best of their ability in accordance with the obligations imposed upon them by law.
LEGAL PROCEEDINGS AND REGULATORY ACTIONS
To the Company’s knowledge, there are no legal proceedings or regulatory actions material to the Company to which it is a party, or has been a party to, or of which any of its property is or was the subject matter, and no such proceedings or actions are known by the Company to be contemplated.
There have been no penalties or sanctions imposed against the Company by a court or regulatory authority, and the Company has not entered into any settlement agreements before any court relating to provincial or territorial securities legislation, or with any securities regulatory authority, in the three years prior to the date of this AIF.
INTEREST OF MANAGEMENT AND OTHERS IN MATERIAL TRANSACTIONS
Other than as disclosed below, and elsewhere in this AIF, no director, executive officer or unitholder or shareholder that beneficially owns, or controls or directs, directly or indirectly, more than 10% of the voting securities of the Company, or any of their respective Associates or affiliates, has any material interest, direct or indirect, in any transaction within the three years before the date of this AIF which has materially affected or is reasonably expected to materially affect the Company or a subsidiary of the Company.
AUDITOR, TRANSFER AGENT AND REGISTRAR
Odyssey Trust Company, at its Calgary, Alberta office, acts as the Company’s transfer agent and registrar and Zeifmans LLP, at its Toronto, Ontario office, acts as the Company’s auditor.
MATERIAL CONTRACTS
Material contracts of the Company, other than contracts entered into in the ordinary course of business, that were entered into within the last financial year or before the last financial year but are still in effect:
a) Warrant Indenture;
b) LPC Purchase Agreement;
c) High Trail Securities Purchase Agreement;
d) High Trail Registration Rights Agreement;
e) Placement Agency Agreement;
f) 2025 Securities Purchase Agreement;
g) 2026 Distribution Agreement; and
h) June 2026 Underwriting Agreement.
The Company’s material contracts described above are filed under the Company’s profile on SEDAR+ at www.sedarplus.ca.
INTERESTS OF EXPERTS
No person or corporation whose profession or business gives authority to a statement made by the person or corporation and who is named as having prepared or certified a part of this AIF or as having prepared or certified a report or valuation described or included in this AIF holds any beneficial interest, direct or indirect, in any securities or property of the Company or of an Associate or affiliate of the Company and no such person is expected to be elected, appointed or employed as a director, senior officer or employee of the Company or of an Associate or affiliate of the Company and no such person is a promoter of the Company or an Associate or affiliate of the Company. Zeifmans LLP is independent of the Company in accordance with the rules of professional conduct of the Institute of Chartered Professional Accountants of Ontario.
AUDIT COMMITTEE
Audit Committee’s Charter
The charter (the “Charter”) of the Company’s Audit Committee is reproduced as Exhibit “A”.
Composition of Audit Committee
As at the date of this AIF, the Audit Committee is composed of Mark Lawson (Chair), Eric Hoskins, Theresa Firestone and Grant Froese, each of whom is a director of the Company.
All of the members of the Audit Committee are “independent” as such term is defined in NI 52-110. The Company is of the opinion that all four members of the Audit Committee are “financially literate” as such term is defined in NI 52-110.
Relevant Education and Experience
All the members of the Audit Committee have the education and/or practical experience required to understand and evaluate financial statements that present a breadth and level of complexity of accounting issues that are generally comparable to the breadth and complexity of issues that can reasonably be expected to be raised by the Company’s financial statements.
Mark Lawson – Mr. Lawson was previously an investment banker with Morgan Stanley in New York where he was involved in the execution of over $6 billion worth of mergers and acquisitions, $8 billion worth of debt offerings and $500 million of equity financings in the healthcare, energy, technology, and media & telecom sector. He received his Bachelor of Arts in Statistical Sciences from The University of Western Ontario, Canada, and his MBA in Finance from The Richard Ivey School of Business, University of Western Ontario, Canada. Mr. Lawson was previously the Chief Financial Officer of a TSX Venture listed company.
Eric Hoskins – Dr. Hoskins served as the Minister of Health for Ontario for 4 years and was responsible for creating, overseeing and administering a $55 billion budget. He was also a member of the Ontario government Cabinet for ten years regularly reviewing and commenting on budgets and financial statements. Dr. Hoskins was the Chief Financial Officer of War Child Canada, a $20 million charity, for 8 years. Dr Hoskins is currently on the audit committee of Canada Health Infowa. He also has a degree in Health Economics and a ICD.D (Institute for Corporate Directors) diploma from the Rotman School of Management.
Theresa Firestone – Ms. Firestone is a senior healthcare executive with over 35 years experience in pharmaceuticals, health & wellness and government and has extensive P&L, strategy development and operations expertise. Ms. Firestone has held executive leadership positions in Canada, Europe and Asia and led teams in 15 different countries. Prior to retirement in 2021, she was Senior Vice President, Health and Wellness at Shoppers Drug Mart (SDM), Canada’s largest retail pharmacy chain. Ms Firestone is and has been a member of audit committees in public and private companies for a number of years and has had overall responsibility for numerous complex businesses including P&Ls, in Canada, Europe and Asia.
Grant Froese – Mr. Froese had a 38-year career with retail giant Loblaw Companies Limited, including 3 years as Chief Operating Officer responsible for all levels of operations and merchandising, as well as oversight of information technology, supply chain, digital/e-commerce, marketing and industry-leading control brands. In his capacity as Chief Operating Officer, Mr. Froese was responsible for financial budgeting, operational P/L and annual revenues of approximately $30 million. Mr. Froese served as Chief Executive Officer of Harvest One Cannabis Inc., where he was responsible for oversight of all aspects of the company’s production, operations and financial matters including, the review and approval of quarterly and annual financial statements, AIF, MD&A, and related corporate disclosures. Mr. Froese has a Diploma in Business Administration.
Audit Committee Oversight
At no time since the commencement of the Company’s most recently completed financial year have any recommendations by the Audit Committee respecting the nomination and/or compensation of the Company’s external auditors not been adopted by the Board.
Pre-Approval Policies and Procedures
Pursuant to the terms of the Audit Committee Charter, the Audit Committee shall pre-approve all non-audit services to be provided to the Company or its subsidiary entities by the Company’s external auditor.
External Auditor Service Fees (By Category)
The aggregate fees billed by the Company’s external auditors during the financial years ended March 31, 2026 and March 31, 2025 were as follows:
| | | | | | | | | | | | | | |
| Financial Period Ending | Audit Fees (CA$)(1) | Audit Related Fees (CA$)(2) | Tax Fees (CA$)(3) | All Other Fees (CA$)(4) |
| 2025 | $380,611 | Nil | $30,100 | Nil |
| 2026 | $540,361 | $8,000 | $70,100 | Nil |
Notes:
(1) “Audit Fees” includes fees necessary to perform the annual audit of the Company’s financial statements. These services include reviewing interim financial statements and disclosure documents related to financings and other attest services required by legislation or regulation, such as comfort letters, consents, reviews of securities filings and statutory audits.
(2) “Audit-Related Fees” include services that are traditionally performed by the auditor.
(3) “Tax Fees” include fees for all tax services other than those included in “Audit Fees” and “Audit-Related Fees”. This category includes fees for tax compliance, tax planning and tax advice. Tax planning and tax advice includes assistance with tax audits and appeals, tax advice related to mergers and acquisitions, and requests for rulings or technical advice from tax authorities.
(4) “All Other Fees” include all other non-audit services, the aggregate fees billed for products and services, other than the services reported under notes (1), (2) and (3) above.
COMPLIANCE PROGRAM
The Company oversees and monitors compliance with applicable laws in each jurisdiction in which it operates. In addition to the Company’s senior executives and the employees responsible for overseeing compliance, the Company has local counsel engaged in every jurisdiction in which it operates and has received legal opinions or advice in each of these jurisdiction regarding (a) compliance with applicable regulatory frameworks, and (b) potential exposure to, and implications arising from, applicable laws in jurisdictions where the Company has operations or intends to operate.
The Company works with third parties who require regulatory licensing in order to handle scheduled drugs. The Company continuously updates its compliance and channel programs to maintain regulatory standards set for drug development. The Company also works with clinical research organizations who maintain batch records and data storage for the Company’s clinical programs.
Additionally, the Company has established a Medical & Clinical Advisory Team, a Research, Clinical and Regulatory Team and a Government Relations and Communications Team with cross-functional expertise in business, neuroscience, pharmaceuticals, mental health and serotonergic agonists to advise management.
In conjunction with the Company’s human resources and operations departments, the Company oversees and implements training on the Company’s protocols. The Company will continue to work closely with external counsel and other compliance experts, and is evaluating the engagement of one or more independent third party providers to further develop, enhance and improve its compliance and risk management and mitigation processes and procedures in furtherance of continued compliance with the laws of the jurisdictions in which the Company operates.
The programs currently in place include monitoring by executives of the Company to ensure that all operations materially conform to and comply with required laws, regulations and operating procedures. The Company is currently in compliance with the laws and regulations in all jurisdictions and the related licencing framework applicable to its business activities.
The Company and, to its knowledge, each of its third-party researchers, suppliers and manufacturers have not received any non-compliance, citations or notices of violation which may have an impact on the Company’s licences, business activities or operations.
The Company conducts due diligence on third-party researchers, medical professionals, clinics, cultivators, processors and others as applicable, with whom it engages. Such due diligence includes but is not limited to the review of necessary licenses and the regulatory framework enacted in the jurisdiction of operation. Further, the Company generally obtains, under its contractual arrangements, representations and warranties from such third parties pertaining to compliance with applicable licensing requirements and the regulatory framework enacted in the jurisdiction of operation.
INSIDER TRADING POLICY AND CODE OF ETHICS AND BUSINESS CONDUCT
Insider Trading Policy
The Company has adopted an insider trading policy to set forth basic guidelines for trading in the Company’s securities (including, without limitation, its Common Shares) to avoid any situation that might have the potential to damage the Company’s reputation or which could constitute a violation of federal or provincial securities law by the Company, its officers, directors, employees, consultants, affiliates and certain family members of such individuals (“Insiders”). Under this policy, Insiders are prohibited from trading in Common Shares and other securities on the basis of material, non-public information relating to the Company until after the information has been disclosed to the public or during a blackout period.
The obligation not to trade on inside information applies not only to the Insiders, but also to persons who obtain such information from Insiders and use it to their advantage. Thus, liability may be imposed upon the Company, its Insiders and also outsiders who are the source of leaks of material information not yet disclosed to the public and the leaks coincide with purchases or sales of the Company’s securities by such insiders, outsiders or by “tippees”.
In order to provide a degree of certainty as to when insider trading is permissible, the policy imposes mandatory blackout periods during the period commencing on the first day following the end of each fiscal quarter or year-end and ending at the close of business on the first trading day following the dissemination by the Company of such quarterly and annual results. In addition, no Insider is permitted to trade any securities of the Company until two trading days after the issuance of any news release in which material information is released to the public. The Company may, from time to time, issue a general blackout period for a specific or indefinite period covering Insiders or specific employees or groups.
The policy also outlines the Company’s reporting obligations for changes in Common Shares owned by Insiders as well as the penalties for violating such policy and applicable laws.
Code of Business Conduct
The Company has adopted a Code of Business Conduct (the “Code”). The Code sets forth standards designed to reasonably deter wrongdoing, promote honest and ethical conduct, promote prompt internal reporting of violations of the Code and promote accountability. All personnel, in discharging their duties, must comply with applicable laws and regulations, the rules of the stock exchange(s) on which the Common Shares are listed as well as the Company’s internal policies.
The Code sets the expectation that personnel learn about laws, rules and regulations that affect what they do at the Company, and raise any questions concerning the applicability, existence or interpretation of any law or regulation or conduct with their supervisor or the legal department of the Company. The Code prohibits personnel from making or participating in making any payments designed to cause or improperly influence the decisions of an individual, a company or a governmental official to act in a way that gives the Company or its personnel an advantage or soliciting, encouraging or actually receiving any bribe or other payment, contribution, gifts or favor that could influence your or another’s decision.
The Code encourages personnel to report any actual or suspected fraud or securities law violations to the Chief Compliance Officer. The Code mandates a safe work environment and a no tolerance policy towards harassment and violence in the workplace. The Code provides guidance on avoiding conflicts of interest and acting in the best interest of the Company. The Code also outlines the requirements or personnel as it relates to disclosure of Company information, confidentiality and maintaining the integrity of the Company’s books and records and intellectual property.
ADDITIONAL INFORMATION
Additional information relating to the Company can be found under the Company’s profile on SEDAR+ at www.sedarplus.ca and on the Company’s website at www.helus.com. Additional information, including directors’ and officers’ remuneration and indebtedness, principal holders of the Company’s securities and securities authorized for issuance under equity compensation plans, will be contained in the Company’s information circular for its most recent annual meeting of shareholders. Additional financial information is provided in the Company’s consolidated financial statements for the most recently completed financial year and the related MD&A.
EXHIBIT “A”
AUDIT COMMITTEE CHARTER
CYBIN INC., dba Helus Pharma
(the “Corporation”)
AUDIT COMMITTEE CHARTER
(Implemented pursuant to National Instrument 52-110 – Audit Committees)
National Instrument 52-110 – Audit Committees (the “Instrument”) relating to the composition and function of audit committees was implemented for reporting issuers and, accordingly, applies to every Cboe Canada listed company, including the Corporation. The Instrument requires all affected issuers to have a written audit committee charter which must be disclosed, as stipulated by Form 52-110F2, in the management information circular of the Corporation wherein management solicits proxies from the security holders of the Corporation for the purpose of electing directors to the board of directors.
This Charter has been adopted by the board of directors in order to comply with the Instrument and to more properly define the role of the Committee in the oversight of the financial reporting process of the Corporation. Nothing in this Charter is intended to restrict the ability of the board of directors or Committee to alter or vary procedures in order to comply more fully with the Instrument, as amended from time to time.
Part 1
Purpose:
The purpose of the Committee is to:
(a) improve the quality of the Corporation’s financial reporting;
(b) assist the board of directors to properly and fully discharge its responsibilities;
(c) provide an avenue of enhanced communication between the directors and external auditors;
(d) enhance the external auditor’s independence;
(e) increase the credibility and objectivity of financial reports; and
(f) strengthen the role of the directors by facilitating in depth discussions between directors, management and external auditors.
1.1Definitions
“accounting principles” has the meaning ascribed to it in National Instrument 52-107 Acceptable Accounting Principles and Auditing Standards;
“Affiliate” means a Corporation that is a subsidiary of another Corporation or companies that are controlled by the same entity;
“audit services” means the professional services rendered by the Corporation’s external auditor for the audit and review of the Corporation’s financial statements or services that are normally provided by the external auditor in connection with statutory and regulatory filings or engagements;
“Charter” means this audit committee charter;
“Committee” means the committee established by and among certain members of the board of directors for the purpose of overseeing the accounting and financial reporting processes of the Corporation and audits of the financial statements of the Corporation;
“Control Person” means any individual or company that holds or is one of a combination of individuals or companies that holds a sufficient number of any of the securities of the Corporation so as to affect materially the control of the Corporation, or that holds more than 20% of the outstanding voting shares of the Corporation except where there is evidence showing that the holder of those securities does not materially affect the control of the Corporation;
“financially literate” has the meaning set forth in Section 1.2;
“immediate family member” means an individual’s spouse, parent, child, sibling, mother or father-in-law, son or daughter-in-law, brother or sister-in-law, and anyone (other than an employee of either the individual or the individual’s immediate family member) who shares the individual’s home;
“independent” means independent only as determined by both the Instrument and the Cboe Canada Listing Manual;
“Instrument” means National Instrument 52-110 – Audit Committees;
“MD&A” has the meaning ascribed to it in National Instrument 51-102;
“Member” means a member of the Committee;
“National Instrument 51-102” means National Instrument 51-102 - Continuous Disclosure Obligations; and
“non-audit services” means services other than audit services.
1.2Meaning of Financially Literate
For the purposes of this Charter, an individual is financially literate if he or she has the ability to read and understand a set of financial statements that present a breadth and level of complexity of accounting issues that are generally comparable to the breadth and complexity of the issues that can reasonably be expected to be raised by the Corporation’s financial statements.
Part 2
2.1 Audit Committee
The board of directors has hereby established the Committee for, among other purposes, compliance with the Instrument.
2.2 Relationship with External Auditors
The Corporation will require its external auditor to report directly to the Committee and the Members shall ensure that such is the case.
2.3 Committee Responsibilities
1.The Committee shall be responsible for making the following recommendations to the board of directors:
(a)the external auditor to be nominated for the purpose of preparing or issuing an auditor’s report or performing other audit, review or attest services for the Corporation; and
(b)the compensation of the external auditor.
2.The Committee shall be directly responsible for overseeing the work of the external auditor engaged for the purpose of preparing or issuing an auditor’s report or performing other audit, review or attest services for the Corporation, including the resolution of disagreements between management and the external auditor regarding financial reporting. This responsibility shall include:
(a)reviewing the audit plan with management and the external auditor;
(b)reviewing with management and the external auditor any proposed changes in major accounting policies, the presentation and impact of significant risks and uncertainties, and key estimates and judgements of management that may be material to financial reporting;
(c)questioning management and the external auditor regarding significant financial reporting issues discussed during the fiscal period and the method of resolution;
(d)reviewing any problems experienced by the external auditor in performing the audit, including any restrictions imposed by management or significant accounting issues on which there was a disagreement with management;
(e)reviewing audited annual financial statements, in conjunction with the report of the external auditor, and obtaining an explanation from management of all significant variances between comparative reporting periods;
(f)reviewing the post-audit or management letter, containing the recommendations of the external auditor, and management’s response and subsequent follow up to any identified weakness;
(g)reviewing interim unaudited financial statements before release to the public;
(h)reviewing all public disclosure documents containing audited or unaudited financial information before release, including any prospectus, the annual report and management’s discussion and analysis;
(i)reviewing the evaluation of internal controls by the external auditor, together with management’s response;
(j)reviewing the terms of reference of the internal auditor, if any;
(k)reviewing the reports issued by the internal auditor, if any, and management’s response and subsequent follow up to any identified weaknesses; and
(l)reviewing the appointments of the chief financial officer and any key financial executives involved in the financial reporting process, as applicable.
3.The Committee shall pre-approve all non-audit services to be provided to the Corporation or its subsidiary entities by the issuer’s external auditor.
4.The Committee shall review the Corporation’s financial statements, MD&A, and annual and interim earnings press releases before the Corporation publicly discloses this information.
5.The Committee shall ensure that adequate procedures are in place for the review of the Corporation’s public disclosure of financial information extracted or derived from the Corporation’s financial statements, and shall periodically assess the adequacy of those procedures.
6.When there is to be a change of auditor, the Committee shall review all issues related to the change, including the information to be included in the notice of change of auditor called for under National Instrument 51-102, and the planned steps for an orderly transition.
7.The Committee shall review all reportable events, including disagreements, unresolved issues and consultations, as defined in National Instrument 51-102, on a routine basis, whether or not there is to be a change of auditor.
8.The Committee shall, as applicable, establish procedures for:
(a)the receipt, retention and treatment of complaints received by the issuer regarding accounting, internal accounting controls, or auditing matters; and
(b)the confidential, anonymous submission by employees of the issuer of concerns regarding questionable accounting or auditing matters.
9.As applicable, the Committee shall establish, periodically review and approve the Corporation’s hiring policies regarding partners, employees and former partners and employees of the present and former external auditor of the issuer.
10.The responsibilities outlined in this Charter are not intended to be exhaustive. Members should consider any additional areas which may require oversight when discharging their responsibilities.
2.4 De Minimis Non-Audit Services
The Committee shall satisfy the pre-approval requirement in subsection (2.3(3)) if:
(a)the aggregate amount of all the non-audit services that were not pre-approved is reasonably expected to constitute no more than five per cent of the total amount of fees paid by the issuer and its subsidiary entities to the issuer’s external auditor during the financial year in which the services are provided;
(b)the Corporation or the subsidiary of the Corporation, as the case may be, did not recognize the services as non-audit services at the time of the engagement; and
(c)the services are promptly brought to the attention of the Committee and approved by the Committee or by one or more of its Members to whom authority to grant such approvals has been delegated by the Committee, prior to the completion of the audit.
2.5 Delegation of Pre-Approval Function
1.The Committee may delegate to one or more independent Members the authority to pre-approve non-audit services in satisfaction of the requirement in subsection (2.3(3)).
2.The pre-approval of non-audit services by any Member to whom authority has been delegated pursuant to subsection (2.5(1)) must be presented to the Committee at its first scheduled meeting following such pre-approval.
Part 3
3.1 Composition
1.The Committee shall be composed of a minimum of three Members.
2.Every Member shall be a director of the issuer.
3.Every Member shall be independent.
4.Every Member shall be financially literate.
5.The board of directors of the Corporation shall appoint or re-appoint the Members after each annual meeting of shareholders of the Corporation.
Part 4
4.1 Authority
Until the replacement of this Charter, the Committee shall have the authority to:
(a)engage independent counsel and other advisors as it determines necessary to carry out its duties;
(b)set and pay the compensation for any advisors employed by the Committee;
(c)communicate directly with the internal and external auditors; and
(d)recommend the amendment or approval of audited and interim financial statements to the board of directors.
Part 5
5.1 Required Disclosure
The Corporation must include in its Annual Information Form the disclosure required by Form 52-110F1.
5.2 Disclosure in Information Circular
If management of the Corporation solicits proxies from the security holders of the Corporation for the purpose of electing directors to the board of directors, the Corporation shall include in its management information circular a cross-reference to the sections in the Corporation’s Annual Information Form that contain the information required by section 5.1.
Part 6
6.1 Meetings
1.Meetings of the Committee shall be scheduled to take place at regular intervals and, in any event, not less frequently than quarterly.
2.Opportunities shall be afforded periodically to the external auditor, the internal auditor and to members of senior management to meet separately with the Members.
3.Minutes shall be kept of all meetings of the Committee.