Exhibit 99.3

SATELLOS BIOSCIENCE INC.
Management’s Discussion and Analysis
For the years ended December 31, 2025, and 2024
DATE OF REPORT: March 27, 2026
Exhibit 99.3
SATELLOS BIOSCIENCE INC.
Management’s Discussion and Analysis
For the years ended December 31, 2025, and 2024
DATE OF REPORT: March 27, 2026
The following discussion is management’s assessment and analysis of the financial performance and financial condition of Satellos Bioscience Inc. (the “Company” or “Satellos”) and should be read in conjunction with the accompanying consolidated financial statements and related notes thereto for the years ended December 31, 2025 and 2024.
Change in Presentation Currency
Effective January 1, 2025, the Company changed its presentation currency from the Canadian dollar (“CA”) to the United States dollar (“USD”). The change in presentation currency was made to better reflect the Company’s business activities and to improve investors’ ability to compare the Company’s financial results with other publicly traded businesses in the industry. For the year ended December 31, 2024, and for all prior periods, we presented our financial statements in CA. The comparative figures disclosed in our consolidated financial statements for the year ended December 31, 2025, and in this MD&A, have been retrospectively changed to reflect the change in presentation currency to the USD, as if the USD had been used as the presentation currency for all prior periods.
All financial information in this MD&A has been prepared in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board (IFRS Accounting Standards) and all dollar amounts are expressed in thousands of USD unless otherwise indicated.
FORWARD-LOOKING STATEMENTS
Certain statements and information in this MD&A contain “forward-looking information” and “forward-looking statements”, within the meaning of applicable Canadian securities laws (collectively herein referred to as “forward-looking statements”). These statements relate to future events or future performance and reflect the Company’s expectations and assumptions regarding the growth, results of operations, performance and business prospects and opportunities of the Company. These forward-looking statements are made as of the date of this MD&A or, in the case of documents incorporated by reference herein, as of the date of such documents. Forward-looking statements are frequently, but not always, identified by words such as “expects”, “expectation”, “anticipates”, “believes”, “intends”, “intention”, “estimates”, “predicts”, “continues”, “potential”, “targeted”, “plans”, “possible”, “goal”, “seek”, “project”, “future”, “likely” and similar expressions, or statements that events, conditions or results “will”, “may”, “could”, “would” or “should” occur or be achieved. Any forward-looking statements or statements of “belief”, including the statements made under “Risks and Uncertainties”, represent the Company’s estimates only as of the date of this MD&A and the documents incorporated by reference herein, respectively, and should not be relied upon as representing the Company’s estimates as of any subsequent date. Forward-looking statements are necessarily based on estimates and assumptions made by Satellos in light of its experience and perception of historical trends, current conditions and expected future developments, as well as factors that Satellos believes are appropriate. Forward-looking statements in this MD&A include, but are not limited to, statements relating to:
| ● | our belief that the Company will be successful in raising additional capital to continue as a going concern; |
| ● | the expected research and development timelines, therapeutic benefits, effectiveness and safety of our product candidates; |
| ● | our belief that the Company’s products and research and development efforts are targeting diseases and conditions with significant unmet medical treatment needs; |
| ● | our belief that the Company has made, and will continue to make progress towards the achievement of certain milestones or objectives; |
| ● | our expectation with respect to meeting milestones and the minimum amount of funds the Company expects to need to raise in order to achieve such milestones and garner additional funding; |
2
| ● | the initiation, timing, cost, progress, outcomes, resource needs and success of our research and development activities, plans and programs; |
| ● | our expectations regarding our ability to design, test and patent novel drug products suitable for advancement into Investigational New Drug (“IND”) enabling studies and clinical trials and the anticipated timelines surrounding such enabling studies; |
| ● | our belief that we will not receive substantive comments from the United States Food and Drug Administration (“FDA”) on our IND applications which unduly delay our plans for and/or timing of a Phase 2 clinical trial in pediatric patients; |
| ● | our expectations that the Notch pathway and AAK1 drug target (both as further described herein) represent drug development opportunities similar or superior to modulation of the epidermal growth factor receptor signaling pathway; |
| ● | our intentions of developing inhibitors to AAK1 (including but not limited to SAT-3247 and SAT-3153) and in showing that such potential inhibitors have desirable effects in relevant models of Duchenne muscular dystrophy (“DMD”) and in other indications, such as other degenerative muscle diseases, muscle injury or trauma, or muscle regeneration generally; |
| ● | our belief that the results of Satellos’ research and development activities, preclinical studies, safety studies or clinical trials have the potential to be commercially competitive with research and development activities, preclinical studies, safety studies or clinical trials conducted by other parties; |
| ● | discoveries we have made in muscle stem cell regulation having the potential to represent insights into a potential root cause of degenerative muscle disorders which has previously not been recognized and which may be therapeutically relevant in the treatment of degenerative muscle disorders; |
| ● | our belief that the Company’s technology can be commercialized, and that such commercialization could be done as effectively or more effectively than other technologies to treat degenerative muscle disorders and conditions or other medical disorders or conditions, or at all; |
| ● | our ability to discover, optimize, select and advance into clinical development therapeutic drug development candidates in a timely, cost-efficient and effective manner, or at all; |
| ● | our ability to translate our discoveries in muscle stem cell regulation into safe and therapeutically effective drug products and the broad applicability of such products; |
| ● | our ability to enter into research and/or commercial development collaborations or partnerships to successfully and profitably advance our drug development candidates; |
| ● | our ability and that of our partners (if any) to advance identified drug development candidates into, and successfully complete, clinical trials; |
| ● | our intention to identify and nominate one or more back-up drug candidates and the potential benefits of having such back-ups; |
| ● | our plans to utilize and deploy MyoRegenXTM in our programs and our continued relationship with OHRI (as defined below); |
| ● | our ability to develop the Company’s novel discoveries into viable therapeutic treatments suitable for clinical development in patients diagnosed with DMD, including, but not limited to, our ability to determine appropriate dosing regimens; |
| ● | the ability of our products to effectively and safely treat Duchenne and other degenerative muscle disorders and conditions or other medical disorders or conditions and the applicability of our products to other disorders and conditions; |
| ● | our expectations regarding future initiation of and enrolment into clinical trials and the timing of future enrolment into clinical trials for our product candidates; |
| ● | our belief that our approach may reduce the risk, time and cost of developing therapeutics by avoiding some of the uncertainty associated with certain research and preclinical stages of drug development; |
| ● | our ability to establish and maintain relationships with collaborators with acceptable preclinical and/or clinical research and development capability, and regulatory and commercialization expertise to enable the development and future commercialization of our technology or products, and the benefits to be derived from such collaborative efforts; |
3
| ● | our ability to enter into agreements or partnerships with pharmaceutical or biotechnology companies that have research and clinical development and/or sales and marketing capabilities and the expected benefits that could be derived therefrom; |
| ● | our ability to generate and protect our potential intellectual property; |
| ● | our ability to operate our business without infringing upon the intellectual property rights of others; |
| ● | our ability to engage third party services with specialized domain expertise for the drafting and submitting of regulatory applications to conduct clinical trials in humans; |
| ● | our ability to establish suitable CMC and GMP protocols as further described herein; |
| ● | the manufacturing capacity of third-party manufacturers for our product candidates; |
| ● | our expectations regarding federal, provincial and foreign regulatory requirements; |
| ● | the timing of, and the costs of obtaining and maintaining, regulatory approvals in the United States, Canada and other jurisdictions; |
| ● | our plans to submit one or more IND applications to and engage in communications with the FDA with the objective of obtaining approvals to initiate clinical trials in patients diagnosed with DMD; |
| ● | the rate and degree of market acceptance and clinical utility of our future products, if any; |
| ● | existing and future corporate alliances and licensing transactions with third parties, and the receipt and timing of any payments to be made by us or to us pursuant to such arrangements; |
| ● | the implementation and execution of our commercial and operational strategy; |
| ● | our ability to engage and retain the consultants or employees required to grow our business; |
| ● | the potential revenue that may be generated from our products, pricing and reimbursement of the patient cost of our drug products by insurers or national health systems, as the case may be, in those jurisdictions where the Company intends to sell its drug products and our ability to achieve profitability; |
| ● | developments relating to our competitors and our industry, including the success of competing therapies that are or become available; |
| ● | the potential growth of the market and demand for our products as well as the estimated pricing and subsequent revenue generation of any potential therapeutics we discover; |
| ● | our belief that any discoveries by the Rudnicki Lab (as defined below) have the potential to have a positive impact on Satellos and our work; |
| ● | our future financial performance, including projected expenditures, future revenue, capital requirements and our needs for additional financing; |
| ● | our belief that our current liquidity position is sufficient to finance our operations for the next 12 months without further financing; |
| ● | our intention to initiate a Phase 2 clinical trial in FSHD (as defined below) and the anticipated timing, enrollment and design of such study; |
| ● | our expectations regarding the conduct and outcomes of our ongoing BASECAMP and TRAILHEAD clinical studies and our plans to expand these studies to additional clinical sites and jurisdictions; and |
| ● | general business and economic conditions and outlook including but not limited to foreign exchange rates and rates of inflation and the evolving regulatory or geo-political landscape. |
Such forward-looking statements reflect our current views with respect to future events, are subject to risks and uncertainties and are necessarily based upon a number of estimates and assumptions that, while considered reasonable by Satellos as of the date of such statements, are inherently subject to significant medical, scientific, business, economic, competitive, political and social uncertainties and contingencies. Many factors could cause our actual results, performance, achievements, prospects or opportunities to be materially different from any future results, performance or achievements that may be expressed or implied by such forward-looking statements. In making the forward-looking statements included in this MD&A, the Company has made various material assumptions, including, but not limited to:
| ● | obtaining positive results from our research and development activities, including clinical trials; |
| ● | our ability to obtain regulatory approvals; |
| ● | assumptions regarding general business, market and economic conditions; |
| ● | assumptions regarding the cost and timing of each study; |
4
| ● | the Company’s ability to successfully advance its preclinical and clinical development programs and execute its plans substantially as currently envisioned; |
| ● | assumptions related to the pricing and reimbursement of its drug products in jurisdictions in which the Company intends to sell its drug products; |
| ● | the Company’s current positive relationships with third parties will be maintained and the potential to develop new partnerships; |
| ● | our ability to continue to use existing licenses for the development of our product(s); |
| ● | the availability (and sources) of financing on reasonable terms; |
| ● | future expenditures to be incurred by the Company, including research and development and operating costs; |
| ● | the Company’s ability to attract and retain skilled consultants and employees; |
| ● | assumptions regarding market competition, market capture and pricing; |
| ● | the products and technology offered by the Company’s competitors; and |
| ● | the Company’s ability to protect patents and proprietary rights. |
In evaluating forward-looking statements, current and prospective shareholders should specifically consider various factors, including the risks outlined under the headings “Foreign Currency Risk”, “Liquidity Risk”, “Credit Risk” and “Risks and Uncertainties” in this MD&A and the risks outlined in the Company’s annual information form for the year ended December 31, 2025 dated March 27, 2026 (the “AIF”). Certain risks and uncertainties that could cause such actual events or results expressed or implied by such forward-looking statements and information to differ materially from any future events or results expressed or implied by such statements and information include, but are not limited to:
| ● | risks related to the early stage of our products; |
| ● | uncertainties related to preclinical product development activities and clinical trial outcomes; |
| ● | uncertainties related to current economic conditions; |
| ● | risks related to rapid technological change; |
| ● | uncertainties related to forecasts and timing of clinical trials and regulatory approval; |
| ● | competition in the market for therapeutic products, including those to treat Duchenne and related diseases; |
| ● | risks related to potential product liability claims; |
| ● | availability of financing and access to capital and the risks associated with the Company’s ability to continue as a going concern; |
| ● | market acceptance and commercialization of products; |
| ● | the availability, costs and supply of materials; |
| ● | risks related to the effective management of our growth; |
| ● | risks related to the reliance on partnerships and licensing agreements; |
| ● | risks related to our reliance on key personnel; |
| ● | risks related to the regulatory approval process for the manufacture and sale of therapeutic products; |
| ● | risks related to the reimbursement process in various jurisdictions where the Company plans to sell its drug products; and |
| ● | our ability to secure and protect our intellectual property. |
The Company cautions that the foregoing list of important factors and assumptions is not exhaustive. Although the Company has attempted to identify on a reasonable basis important factors and assumptions related to forward-looking statements, there can be no assurance that forward-looking statements will prove to be accurate, as events or circumstances or other factors could cause actual results to differ materially from those estimated or projected and expressed in, or implied by, these forward-looking statements. Other than as specifically required by law, the Company undertakes no obligation to update any forward-looking statement to reflect events or circumstances after the date on which such statement is made, or to reflect the occurrence of unanticipated events, whether as a result of new information, future events or results or otherwise. Accordingly, readers should not place undue reliance on forward-looking statements.
5
NATURE OF BUSINESS AND OVERVIEW OF OPERATIONS
Overview of the Business
Satellos is a publicly listed (NASDAQ: MSLE, TSX: MSCL), clinical-stage drug development company focused on restoring natural muscle repair and regeneration in degenerative muscle diseases. Through its research, Satellos has developed SAT-3247, a first-of-its-kind, orally administered small molecule drug designed to address deficits in muscle repair and regeneration. SAT-3247 targets AAK1, a key protein that Satellos has identified as capable of helping restore muscle stem cell signaling that is disrupted in DMD. By addressing the loss of dystrophin-dependent cues, SAT-3247 may re-establish the signals that support effective muscle regeneration. SAT-3247 is currently in clinical development as a potential disease-modifying treatment, initially for DMD. Satellos is also working to identify additional muscle diseases or injury conditions where restoring muscle repair and regeneration may have therapeutic benefit and represent future clinical development opportunities.
Satellos Bioscience Inc. was incorporated under the Canada Business Corporations Act on July 27, 2012 and commenced trading on the Toronto Stock Exchange (the “TSX”) on February 14, 2024, under the symbol “MSCL”, and the Nasdaq Global Market (“Nasdaq”) under the trading symbol “MSLE” on February 6, 2026.
As at December 31, 2025, the Company had two wholly owned subsidiaries, Satellos Bioscience Australia Pty Ltd. (an entity incorporated under the laws of Australia) and Satellos Bioscience US, Inc. (incorporated under the laws of Delaware, USA).
The Company’s head office is located at Royal Bank Plaza, South Tower, 200 Bay St., Suite 2800, Toronto, Ontario, M5J 2J1, and the Company’s registered and records office is located at Royal Bank Plaza, South Tower, 200 Bay St., Suite 2800, Toronto, Ontario, M5J 2J1.
Achievements and Highlights in the year ended December 31, 2025
On February 10, 2025, the Company announced that the single-ascending dose (“SAD”) cohorts, four multiple-ascending dose (“MAD”) cohorts, and one cross-over food effect single-dose cohort of the Company’s Phase 1a clinical trial with SAT-3247 had been fully enrolled.
On March 19, 2025, the Company announced initial safety and pharmacokinetic (“PK”) data of SAT-3247 from the Phase 1a clinical trial in an oral presentation at the 2025 Muscular Dystrophy Association (MDA) Clinical & Scientific Conference.
The purpose of the Phase 1a clinical study was to assess the safety, tolerability and PK of SAT-3247. In Phase 1a, 72 healthy volunteers were randomized across five SAD cohorts (including one cross-over food effect cohort) with single oral doses of up to 400 mg, and four MAD cohorts with daily oral doses up to 240 mg/day for 7 consecutive days. Phase 1a data showed that:
| ● | SAT-3247 was safe and well tolerated across all healthy volunteer cohorts. At predicted human efficacious dose levels (i.e., between 50 and 150 mg total daily dose), SAT-3247 did not display adverse clinical findings on any parameter measured, including clinical labs, vital signs, ECG’s, and physical exams. No moderate or greater drug-related adverse events were reported at any dose studied and any mild events reported were reversible. |
| ● | Phase 1a PK data demonstrated consistency with results from the Company’s preclinical studies. These PK results confirmed post-dose plasma concentrations of SAT-3247 were sustained at levels and time courses which the Company’s research findings suggest are most likely to yield a therapeutic effect on muscle regeneration and strength. |
6
On May 22, 2025, the Company announced data from a Phase 1b clinical study with SAT-3247. The Phase 1b was designed as an open-label study in which five adult male DMD patients, ages 20 – 27, were enrolled and treated with SAT-3247 over a period of 28 days. The results from this study were further updated at the 30th Annual Congress of the World Muscle Society as described below. Study participants were offered the option to enroll into an 11-month, long-term follow-up study with SAT-3247 and the first patient in this study was dosed in October 2025 (LT-001 or TRAILHEAD).
On June 18, 2025, the Company announced the election of Iris Loew-Friedrich, M.D., Ph.D., and Selwyn Ho, MBBS, to its Board of Directors. As part of the Annual General Meeting, Rima Al-awar, Ph.D., William Jarosz, J.D., and William McVicar, Ph.D., did not stand for re-election.
On July 16, 2025, the Company announced the appointment of Wildon Farwell, M.D., MPH, as Chief Medical Officer. Dr. Farwell joined Satellos from Dyne Therapeutics (Nasdaq: DYN), where he most recently served as CMO and medical advisor. At Dyne, Dr. Farwell built the development organization, led the protocol development and regulatory submissions for their DMD and myotonic dystrophy type 1 (DM1) programs, oversaw the conduct of multiple potentially registrational clinical studies, and contributed to several successful capital raises. Before joining Dyne, he spent a decade at Biogen in increasing leadership roles, including on clinical programs resulting in the approval of Spinraza and Qalsody as then-novel medicines for the treatment of the neuromuscular diseases SMA and ALS, respectively. Prior to moving into industry, Dr. Farwell served as an assistant professor of medicine at Harvard Medical School and was a physician at Brigham and Women’s Hospital and the VA Boston Healthcare System. He earned his medical degree from the University of Missouri School of Medicine and holds a Master of Public Health in clinical effectiveness from the Harvard T.H. Chan School of Public Health.
The Company announced on September 22, 2025, that 1.7 million warrants with an exercise price of CA$0.60 had been exercised for gross proceeds of CA$1.0 million.
On September 22, 2025, the Company announced the submission of an IND application to the U.S. FDA, along with parallel regulatory filings in the United Kingdom, Europe, Serbia and Australia, to initiate a Phase 2 clinical trial of SAT-3247 in ambulatory children with DMD.
On October 10, 2025, at the 30th Annual Congress of the World Muscle Society in Vienna, Austria, the Company announced new clinical trial data from the Company’s Phase 1b clinical trial of SAT-3247 in adults (aged 20-27 years) with DMD further demonstrating potential tolerability and providing a preliminary indication of potential for therapeutic efficacy of SAT-3247 following treatment over a period of 28 days.
Individuals treated with SAT-3247 over a 28-day period demonstrated an increase in grip strength far greater than seen in the Duchenne natural history in this age group. Specifically, a 118.6% mean improvement in maximum grip strength was observed in the dominant hand and 97.9% mean improvement in the non-dominant hand, representing an approximate doubling of grip strength from ~2 kg to ~4 kg. These improvements are inconsistent with published natural history and were correlated with higher drug concentrations on Day 15 and higher baseline creatinine (a surrogate for increased muscle mass), which we believe indicates our drug is having the desired impact on muscle. The increases in grip strength seen in 3 of the 5 patients after 28 days appear to be moving into “white space”, strength measures not seen in the natural history for patients in this age demographic. Improvements of this nature (if sustained) may provide an opportunity for early approval consideration of SAT-3247.
Furthermore, participants exhibited a 5.8% mean improvement of predicted forced vital capacity; an increase that is also inconsistent with natural history with declines about 5% annually among adults with Duchenne. All other measures remained stable over the study period. No drug-related adverse events of moderate severity or higher were observed in either study, and no dose-limiting toxicities occurred.
7
Data presented at the meeting also demonstrated that SAT-3247 was safe and well-tolerated with a desirable pharmacokinetic (PK) profile across the Phase 1a portion of the study conducted in 72 healthy adult human volunteers.
On October 21, 2025, the Company announced that the first patient had been dosed in the Company’s open-label, long-term follow-up study (LT-001 or TRAILHEAD) involving 11 additional months of SAT-3247 treatment of adult DMD patients previously treated in the Phase 1b study noted above.
On November 14, 2025, the Company announced that it had appointed Mark Nawacki, co-founder, president and former CEO of Searchlight Pharma, to its board of directors.
On December 9, 2025, the Company announced that it received IND clearance by the FDA, as well as other global regulatory agencies, to conduct SAT-3247-CL-201 (aka BASECAMP), a three-month, randomized, double-blind, placebo-controlled, proof-of-concept, Phase 2 study of SAT-3247 in 51 ambulatory children with DMD. In addition to the FDA clearance, the United Kingdom’s Medicine and Healthcare products Regulatory Agency granted authorization of the Company’s Clinical Trial Application (“CTA”); Australia’s Human Research Ethics Committee accepted the Therapeutic Goods Administration’s Clinical Trial Notification scheme for regulatory authorization; and the Medicines and Medical Devices Agency of Serbia approved the CTA. On December 12, 2025, the CTA was approved in Canada and on January 22, 2026, the CTA was approved by the European Union. The Company is currently cleared to proceed with 60mg and 120mg doses of SAT-3247 in the UK, Australia, Serbia and the European Union. The FDA and Health Canada cleared the Company to proceed with the 60mg dose but require the Company to provide additional pharmacokinetic data in pediatric patients before being cleared to proceed with the 120mg dose.
Subsequent Events
On January 27, 2026, the Company completed a consolidation of its outstanding common shares (“Common Shares”) on the basis of one post-consolidation Common Share for every 12 pre-consolidation Common Shares (the “Consolidation”). The Consolidation took effect on the TSX at market open on January 30, 2026.
On January 29, 2026, the Company announced the appointment of Antoinette Paone as Chief Development Officer and Head of Regulatory Affairs. Ms. Paone brings extensive experience leading regulatory strategy from clinical development through approval, including her work on Kalydeco and Orkambi at Vertex Pharmaceuticals (Nasdaq: VRTX). She joins Satellos from Generation Bio (Nasdaq: GBIO), where she most recently served as Chief Operating Officer.
On February 6, 2026, the Common Shares began trading on Nasdaq.
On February 9, 2026, the Company announced that it had completed an underwritten public offering of 5,168,019 Common Shares, which included the exercise of the underwriters’ option to purchase an additional 712,574 Common Shares and, in lieu of Common Shares for certain investors, pre-funded warrants to purchase 495,049 Common Shares. The Common Shares were sold at a price of US$10.10 per share (C$13.81 per Common Share) and the pre-funded warrants were sold at a price of US$10.09999 per pre-funded warrant (C$13.80999 per pre-funded warrant), which represents the per share price for the Common Shares less the C$0.00001 per share exercise price for each pre-funded warrant. The total gross proceeds to the Company were approximately US$57,200, before deducting the underwriting discounts and commissions.
On February 12, 2026, the Company announced that the first participant had been dosed in BASECAMP, a 51 patient, three-month, randomized, double-blind, placebo-controlled, proof-of-concept, Phase 2 pediatric study of SAT-3247 for DMD.
On March 10, 2026, the Company announced interim clinical and biomarker data for SAT-3247 at the Muscular Dystrophy Association (“MDA”) Clinical & Scientific Conference. The data include interim observations from
8
the ongoing TRAILHEAD study in adults with DMD serum proteomic analysis from the previously completed 28-day, CL-101 Phase 1a/b trial, and the development of a novel muscle regeneration assessment tool. The Company also presented preclinical findings demonstrating enhanced muscle strength in a mouse model of FSHD.
Description of Business Strategy and Programs
The Company’s primary goal is the development of disease modifying therapeutic drugs for the treatment of severe muscle conditions of unmet medical need. Our core technology is based on discoveries by the Company’s scientific founder and Chief Discovery Officer, Dr. Michael Rudnicki, in understanding and modulating muscle stem cell function and its role in muscle regeneration. Multiple peer reviewed publications from Dr. Rudnicki’s lab (the “Rudnicki Lab”) at the Ottawa Hospital Research Institute (the “OHRI”) have advanced the understanding of the identity and behavior of muscle stem cells including their role in health and disease. For instance, the Rudnicki Lab was the first to define so called muscle stem cells (a.k.a. ‘satellite stem cells’) and characterize a sub-population as bona fide multipotent stem cells capable of both self-renewal and regeneration (Source: Kuang et al., 2007, Cell). Dr. Rudnicki was also first to demonstrate that such stem cells exist as a special body of cells capable of regeneration, and subsequently elucidate their biological mechanism of action and identify means to modulate their activity. He further linked deficiencies in muscle stem cell function directly to the pathology of Duchenne as a potential causal factor in the progressive muscle destruction that occurs in this lethal disease (Source: Dumont et al., 2015, Nature Medicine).
The basic principle governing how muscle stem cells contribute to the creation of new muscle cells and hence muscle regeneration, through a process known as asymmetric division, is depicted below in Figure 1.
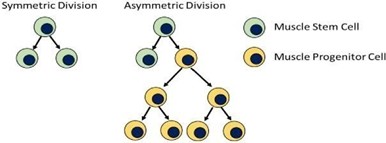
Figure 1: Muscle stem cells undergo asymmetric divisions in response to injury stimuli. Muscle
progenitor cells are generated to produce new muscle tissue or repair injured muscle.
Asymmetric muscle stem cell divisions result in one stem cell being produced and one progenitor muscle cell. The former maintains the pool of stem cells to be called on to respond to future injury. Progenitor muscle cells by contrast, continue to replicate through normal cell mitosis to generate potentially thousands of cells that ultimately incorporate into and become functional muscle tissue. Findings from the research of Dr. Rudnicki have linked deficits in asymmetric division, to the progressive muscle loss which is a principal pathology of DMD and other degenerative diseases.
To apply our understanding of muscle regeneration to therapeutic development in degenerative muscle conditions or disorders, Satellos employs a proprietary discovery platform developed by the Rudnicki laboratory at OHRI called, MyoReGenX™. An automated microscopy system, MyoReGenX™ recapitulates the muscle stem cell environment ex-vivo (i.e., outside the body) and enables Satellos to identify and assess opportunities for developing novel therapeutic treatments.
9
Lead Development Program: Duchenne
The Company’s first application of its technology is directed towards the discovery and development of a small molecule drug for the treatment of Duchenne, the most common fatal genetic disorder diagnosed in childhood affecting approximately one in 4,000 male births per year, worldwide. As depicted in the below Figure 2, individuals living with Duchenne experience severe and progressive loss of muscle function during their lives, often exhibited by loss of ambulation before their teenage years and generally culminating in death before the end of their third decade of life. There is no known cure.
Despite this dire scenario, Satellos takes hope for its novel approach from the fact that individuals living with Duchenne do make functional muscle as young children, albeit not as effective as their healthy peers. Our interpretation of the progressive nature of Duchenne, also depicted in Figure 2, is that the unmistakable signs of motor impairment and ambulatory challenges that become apparent during childhood represent a ‘tipping point’ in the balance between muscle damage and repair where regeneration fails to keep up with damage. Satellos has designed SAT-3247 with the goal of resetting the balance of regeneration over degeneration by enhancing the process of asymmetric division and the ensuing creation of new muscle cells.
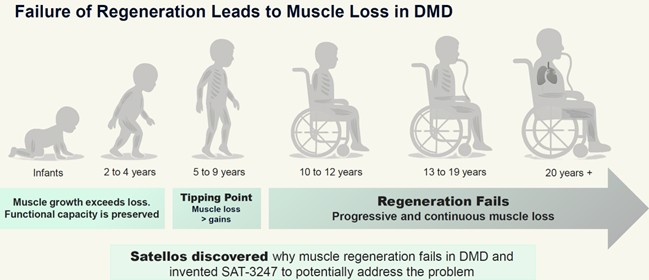
Figure 2: Progressive muscle loss a hallmark of Duchenne muscular dystrophy
Duchenne is caused by a mutation in the dystrophin gene that results in impairment to or loss of the dystrophin protein. Dr. Rudnicki demonstrated that muscle stem cells require a signal from the dystrophin protein to properly and efficiently divide in an asymmetric fashion (Source: Dumont et al. 2015, Nature Medicine.). As described in Figure 3 below, without the dystrophin signal, muscle stem cells fail to divide efficiently, often creating copies of themselves rather than making the progenitor cells needed to create new muscle.
10
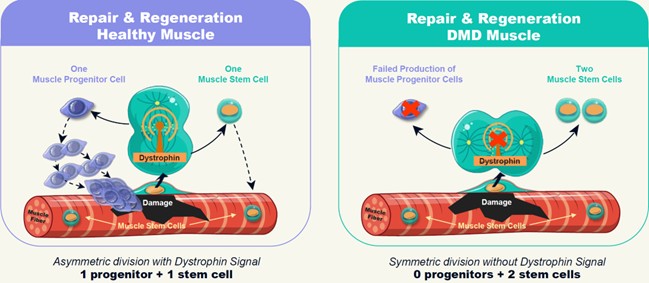
Figure 3: Imbalanced Stem Cell Division
To address this problem, Satellos’ therapeutic strategy is to restore the missing signaling role of dystrophin by drug treatment - thereby resetting the muscle regeneration process.
Restoring the Missing Dystrophin Signal via AAK1 Inhibition
Deploying MyoReGenX™ to build on the identification and discovery of this previously unreported signaling role of dystrophin, in collaboration with the Rudnicki lab, Satellos undertook a systematic assessment, evaluation and prioritization of molecular pathways for their potential to safely rescue asymmetric stem cell divisions in the absence of dystrophin. From this exercise conducted over a multi-year period, the Company identified and selected Adaptor Associated Kinase 1 (aka “AAK1”), a protein kinase in the molecular signaling pathway known as “Notch”. Satellos has generated extensive preclinical data in the Mdx mouse, a gold standard research model bearing the same genetic defect as patients with Duchenne, demonstrating that treatment of these research mice through inhibition of AAK1 with SAT-3247 has potential to restore the process of asymmetric division in muscle stem cells. Our preclinical studies have further shown that inhibition of AAK1 with SAT-3247 enables muscle regeneration with the potential to increase muscle strength. Thus, we believe, SAT-3247 represents a potential novel therapeutic drug for the treatment of Duchenne in humans. Figure 4 below depicts our understanding of the mechanism by which our lead drug candidate, SAT-3247, affects asymmetric division and the muscle stem cell mediated regeneration via inhibition of AAK1.
11
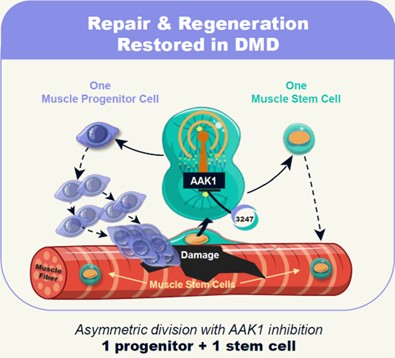
Figure 4: Satellos Approach: Reset regeneration with SAT-3247
Small molecule inhibitors of AAK1 have previously been described for non-muscle related disease indications by Lexicon Pharmaceuticals Inc., an unrelated biotech company, which has reported what appears to be acceptable safety profiles in multiple human clinical trials spanning hundreds of patients. We believe this provides some initial indications of the potential safety of AAK1 inhibition.
Satellos announced positive preclinical data presented at the March 2024 MDA Clinical and Scientific Conference showing that SAT-3247 can improve skeletal muscle function in multiple mouse models of muscle degeneration. The preclinical data presented show the broad potential of SAT-3247 to improve skeletal muscle function as it has been demonstrated in three mouse models of muscle degeneration: mdx model of Duchenne, FLExDUX4 model of FSHD, and a muscle injury model in wildtype mice. In all instances, treatment with SAT-3247 over a three-to-four-week period resulted in a statistically significant improvement in muscle force versus animals receiving placebo.
In October 2024, Satellos announced data presented at the 29th Annual Congress of the World Muscle Society in Prague. The data presented from the open-label pilot study demonstrated that treatment of two DMD canines with SAT-3247 improved measures of strength to near normal levels.
The Company has filed for patent protection on SAT-3247 and other inhibitors of AAK1. Please refer to the Intellectual Property section in the AIF for further details on its intellectual property strategy and filings and its licensing agreement with the OHRI.
Regulatory Designations for SAT-3247
The FDA granted both Orphan Drug Designation and Rare Pediatric Disease Designation to SAT-3247 for the potential treatment of DMD. Orphan Drug Designation applies to therapies targeting rare diseases affecting fewer than 200,000 people in the U.S. and provides benefits including seven-year market exclusivity upon
12
approval, exemption from FDA application fees, tax credits for clinical trials, and eligibility for a priority review voucher.
The Rare Pediatric Disease Designation specifically supports treatments for serious and life-threatening conditions primarily affecting children under 18 years old. Under this program, drug sponsors may qualify for a priority review voucher upon approval, which can be used to accelerate the review of a future marketing application for another product or sold to another sponsor. These designations recognize the unmet medical need in DMD and provide regulatory and financial incentives to support SAT-3247’s development.
Clinical Development of SAT-3247
The Company has advanced SAT-3247 through IND enabling studies, GMP manufacturing, a Phase 1a clinical study in 72 healthy adult volunteers and a Phase 1b clinical study in 5 adult DMD patients.
In September 2024, the first participant in the first-in-human Phase 1a clinical trial was dosed in Australia following regulatory approvals. The trial consisted of two components: a randomized, placebo-controlled study in healthy volunteers evaluating safety and PK across multiple dose cohorts, and an open-label study in adults with genetically confirmed DMD examining safety, PK, and potential pharmacodynamic markers. The healthy volunteer portion enrolled 72 participants across single ascending dose, multiple ascending dose, and food effect cohorts, while the DMD component included 5 participants receiving a single dose daily of SAT-3247 over 28 days.
In February 2025, the Company announced that the SAD, MAD and food effect dose cohorts of the Phase 1 clinical trial had been fully enrolled and in March 2025, the Company announced initial Phase 1 data in an oral presentation at the 2025 MDA conference as described above.
In May 2025, Satellos announced Phase 1b data, from its open-label study, demonstrating early signs that SAT-3247 may have the potential to positively affect grip strength in a statistically significant manner. This data was further updated at the World Muscle Society Conference in October 2025 as described above.
The TRAILHEAD study, a long-term follow-up study for the patients who completed the Phase 1b study was initiated in Q3 2025 with the first patient dosed in October 2025.
This study will evaluate, over an eleven-month period, longer-term safety, changes in muscle composition by MRI, and functional outcomes including grip strength and FVC. The design allows patient assessments and reporting every three months during treatment. The Company is working to expand the TRAILHEAD protocol to include additional participants in Australia and plans to open the study in the U.S. — both subject to regulatory and clinical site approvals.
The Company filed IND and equivalent applications to the relevant regulatory authorities in the USA, the UK, the EU, Australia, and Serbia in September 2025 seeking approvals to conduct the BASECAMP study, a global, randomized, placebo-controlled Phase 2 clinical trial in ambulatory pediatric DMD patients.
The BASECAMP study will evaluate SAT-3247 in 51 ambulatory children with DMD aged 7, 8 or 9 years. Primary endpoints include safety, tolerability and effect on muscle force. Secondary endpoints will assess SAT-3247’s impact on muscle quality, function and regeneration. The BASECAMP trial is actively enrolling, and Satellos plans to establish 25 sites for the study at clinical centers in the United States, Europe, the United Kingdom, Australia, Canada and Serbia.
Please refer to the section “Regulatory Process” in the Company’s AIF for further details on the clinical drug development process.
13
Follow-On Program
There are more than 30 types of muscular dystrophy that affect humans. Each of these dystrophies has different causes that manifest into conditions ranging in severity from benign, small impairments to motor function, to the full loss of ambulation, or even death. Satellos has conducted proof of concept preclinical studies in relevant animal disease models showing potential for benefit by restoring the muscle regeneration process in Lama-2 Related Muscular Dystrophy (prevalence estimates between one in 50,000 and one in 400,000 births), Collagen-VI Related Muscular Dystrophy (prevalence of severe form of the disease estimated to be one in 1,000,000 births) and Facioscapulohumeral muscular dystrophy (“FSHD”) (prevalence of 4 per 100,000 individuals). These represent potential follow-on disease indications or programs for Satellos to consider in the future. The Company also plans to evaluate additional dystrophies as part of its ongoing research and development efforts.
The Company intends to initiate a Phase 2 clinical trial in FSHD. The planned objectives and endpoints of the study would include safety, drug concentration, fluid-based biomarkers and efficacy. The Company anticipates that the study would enroll approximately 50 adult participants with FSHD in a three-month placebo-controlled study with a nine month long term extension. The Company currently anticipates filing the necessary regulatory documents to initiate a Phase 2 clinical trial in the United States and Canada in the second quarter of 2026.
REVIEW OF FINANCIAL RESULTS
All tabular amounts below are presented in thousands of US dollars, except for per share amounts.
On January 27, 2026, the Company completed a twelve-for-one share consolidation of its issued and outstanding Common Shares. In accordance with IFRS Accounting Standards, the weighted average number of common shares outstanding and the basic and diluted net loss per share for all years presented has been retrospectively adjusted to reflect the consolidation.
The financial information reported herein was derived from the audited annual consolidated financial statements for the years ended December 31, 2025 and 2024.
Selected Financial Information
| | 2025 | | 2024 |
| | $ | | $ |
Research & development expenses | | 18,426 | | 14,359 |
General & administrative expenses |
| 8,033 |
| 6,039 |
Other (income)/expense |
| (1,758) |
| 236 |
Income tax expense |
| 172 |
| — |
Net loss |
| (24,873) |
| (20,634) |
Basic and diluted net loss per Common Share |
| (1.70) |
| (2.16) |
Total assets |
| 31,889 |
| 50,747 |
Total non-current financial liabilities |
| — |
| — |
We have not earned revenue in any of the previous fiscal years.
For the year ended December 31, 2025, we reported a net loss of $24,873 ($1.70 loss per Common Share), compared to a net loss of $20,634 ($2.16 loss per Common Share) for the year ended December 31, 2024. The $4,239 increase in net loss for the year ended December 31, 2025, compared with the year ended December 31, 2024, was primarily a result of increased Research and Development (“R&D”) expenses related to clinical activities associated with SAT-3247, particularly initial and setup costs for the BASECAMP study and the ongoing TRAILHEAD study initiated in the fourth quarter of 2025. General and administrative (“G&A”) expenses also increased for the year ended December 31, 2025, compared with the year ended December 31, 2024 due to additional personnel
14
fees and professional fees to support advancing operations and reporting requirements in the current year. In addition, for the year ended December 31, 2024, other expense included an impairment loss of $2,905 to fully write down the remaining carrying value of an intangible asset which offset finance income and foreign currency gains.
Total assets decreased to $31,889 at December 31, 2025 from $50,747 at December 31, 2024, primarily due to the use of cash to fund the Company’s expanding clinical trial activities. Cash and short-term investments declined as funds were deployed to support research and development activities, including the completion of the Phase 1 clinical program, the initiation of the TRAILHEAD study, and the setup of the BASECAMP study for SAT-3247.
Results of Operations for the Years ended December 31, 2025, and 2024
Research and development expenses:
| | Year ended | | Year ended |
| | December 31, 2025 | | December 31, 2024 |
| | $ | | $ |
Salaries and management fees | | 3,359 | | 2,687 |
Discovery expenses |
| 682 |
| 698 |
Preclinical expenses |
| 2,160 |
| 5,420 |
Chemistry, manufacturing and controls |
| 1,117 |
| 2,495 |
Clinical expenses |
| 9,600 |
| 2,459 |
Stock-based compensation |
| 1,508 |
| 600 |
Total research and development expenses |
| 18,426 |
| 14,359 |
Research and development expenses increased by $4,067 to $18,426 for the year ended December 31, 2025, compared to $14,359 for the year ended December 31, 2024. Factors contributing to the increase in R&D expenses in the current year period are primarily the result of the following:
| ● | Salaries and management fees increased by $672 for the year ended December 31, 2025, compared with the prior year. The increase is mainly related to higher headcount in the current year to support expanded clinical activities. |
| ● | Preclinical expenses decreased by $3,260 in the current year period. The preclinical expense in the current period is related to long-term toxicology work to support clinical development. In the comparative period, preclinical expenses were related to IND enabling studies conducted to support the necessary regulatory filing, and to initiate clinical development of SAT-3247. |
| ● | Chemistry, and manufacturing controls (“CMC”) expenses decreased by $1,378 as compared to the prior year period. CMC activities in the comparative period related to the process development and manufacturing of SAT-3247 for clinical use, while current period costs decreased due to a more focused scope of process development activities. |
| ● | Clinical expenses increased by $7,141 for the year ended December 31, 2025. Clinical costs incurred in the current period are associated with the completion of the Phase 1 healthy volunteer clinical study and the Phase 1b clinical study in adult DMD patients, the ongoing TRAILHEAD study, for which patients were dosed in the fourth quarter, and the initiation and setup costs for the BASECAMP study, which also initiated in Q4 2025. Clinical costs incurred in the comparative period were related to costs associated with initiating and conducting the Phase 1 healthy volunteer clinical study. |
| ● | Non-cash stock-based compensation increased by approximately $908 for the year ended December 31, 2025 compared with the year ended December 31, 2024 due to new grants issued in the first half of 2025 and the timing of the related vesting. |
15
General and administrative:
| | Year ended | | Year ended |
| | December 31, 2025 | | December 31, 2024 |
| | $ | | $ |
Salaries and management fees | | 3,337 |
| 2,723 |
Professional fees | | 2,374 |
| 1,688 |
Other operating expenses | | 590 |
| 572 |
Stock-based compensation | | 1,727 |
| 1,041 |
Depreciation | | 5 |
| 15 |
Total general and administrative expenses | | 8,033 |
| 6,039 |
General and administrative expenses increased by $1,994 to $8,033 for the year ended December 31, 2025, as compared to $6,039 for the year ended December 31, 2024. Changes to the components for general and administrative expenses presented in the table above are primarily the result of the following:
| ● | Salaries and board fees increased by $614 in the year ended December 31, 2025, compared with the prior year. The increases are primarily related to salary and board fee adjustments, increased staffing to support expanded operations and a separation payment in the current year. |
| ● | Professional fees increased by $686 in the year ended December 31, 2025, compared with the prior year. The increase is primarily related to legal and audit fees, and fees associated with public company reporting obligations. |
| ● | Non-cash stock-based compensation increased by $686 for the year ended December 31, 2025, compared with the prior year due to new grants issued in 2025 and the timing of the related vesting. |
Other income and expenses:
| | Year ended | | Year ended |
| | December 31, 2025 | | December 31, 2024 |
| | $ | | $ |
Finance income | | 1,360 |
| 1,003 |
Loss on derivative financial instruments | | — |
| (3) |
Impairment of intangible assets | | — |
| (2,905) |
Foreign exchange gain | | 398 |
| 1,669 |
Total other income/(expenses) | | 1,758 |
| (236) |
Other income and expenses were a net income of $1,758 in the year ended December 31, 2025, compared to net expenses of $236 in the prior year. Changes to the components for other income and expenses presented in the table above are primarily the result of the following:
| ● | Finance income increased in the current year by $357 as compared to the prior year, related to interest earned on cash and cash equivalents and short-term investments from an increased average balance. |
| ● | In the year ended December 31, 2024, we recorded an impairment loss of $2,905, to fully write down the remaining carrying value of an intangible asset. |
| ● | The foreign exchange gain in the year ended December 31, 2025 of $398, decreased as compared to a foreign exchange gain of $1,669 in the prior year. The decrease is primarily related to the change in the functional currency of the Company from Canadian dollars to US dollars effective January 1, 2025. |
16
Summary of Quarterly Results
The table below is derived from unaudited quarterly results and was prepared by management for the eight previous quarters to December 31, 2025.
| | Q4 2025 | | Q3 2025 | | Q2 2025 | | Q1 2025 | | Q4 2024 | | Q3 2024 | | Q2 2024 | | Q1 2024 |
| | $ | | $ | | $ | | $ | | $ | | $ | | $ | | $ |
R&D expenses |
| 5,455 |
| 3,994 |
| 4,435 |
| 4,542 |
| 3,999 |
| 2,387 |
| 3,567 |
| 4,406 |
G&A expenses |
| 2,192 |
| 1,972 |
| 1,932 |
| 1,937 |
| 1,678 |
| 1,313 |
| 1,326 |
| 1,722 |
Other (income) and expenses |
| (402) |
| (164) |
| (791) |
| (401) |
| (1,221) |
| 2,924 |
| (469) |
| (998) |
Income taxes |
| 58 |
| 19 |
| 32 |
| 63 |
| — |
| — |
| — |
| — |
Net Loss |
| (7,303) |
| (5,821) |
| (5,608) |
| (6,141) |
| (4,456) |
| (6,624) |
| (4,424) |
| (5,130) |
Loss per Common Share |
| (0.47) |
| (0.36) |
| (0.36) |
| (0.48) |
| (0.44) |
| (0.72) |
| (0.48) |
| (0.60) |
Loss per Common Share amounts presented in the summary of quarterly results have been retrospectively adjusted to reflect the Consolidation, which occurred subsequent to the reporting date but prior to the date of this MD&A.
R&D expenses increased in the year ended December 31, 2025 as compared to the prior year, primarily due to clinical costs related to the completion of the Phase 1 clinical program, initiation of the BASECAMP study, and the ongoing TRAILHEAD study. R&D expenses in Q3 2025 were lower than prior quarters in the current year ended December 31, 2025, primarily as a result of the recognition of a R&D tax incentive credit that was applied against R&D expenditures in Q3 2025. In 2024, R&D expenses reflected the completion of the IND enabling studies necessary for the IND submission in Q2 2024 and clinical costs on Phase 1 clinical programs for which the first patient was dosed in Q3 2024.
G&A expenses have increased in the year ended December 31, 2025 as compared to the prior year primarily related to increased personnel expenses, non-cash stock-based compensation expenses and professional fees associated with the Nasdaq listing.
Beginning in Q1 2025, other income and expenses primarily reflect foreign exchange gains and losses on the Company’s cash and cash equivalents held in CA and interest income earned on investments. Between Q1 2024 and Q4 2024, excluding Q3 2024, other income and expenses primarily reflect foreign exchange gains and losses on the Company’s cash and cash equivalents and investments held in USD and interest income earned on short-term investments.
Net loss increased in Q3 2024, because the Company recognized an impairment of $2,905 to fully write down the remaining carrying value of an intangible asset.
Quarter ended December 31, 2025, compared to quarter ended December 31, 2024
The net loss for the three-months ended December 31, 2025, was $7,303 as compared with a net loss of $4,456 for the three-months ended December 31, 2024. The increase in net loss was primarily a result of increased R&D activities associated with the advancement of our clinical programs. In addition, net loss was further increased due to lower other income when compared to the prior year period due to foreign exchange impacts from the change in the functional currency from Canadian dollars to US dollars effective in the current year.
Research and development expenses increased by $1,456 to $5,455 for the three-months ended December 31, 2025, as compared to $3,999 for the three-months ended December 31, 2024. The increase primarily reflects higher R&D costs related to the initiation and setup of the BASECAMP study and the ongoing TRAILHEAD study, with first patient dosed in the fourth quarter of the current year.
17
General and administrative expenses increased by $514 to $2,192 for the three-months ended December 31, 2025, as compared with $1,678 for the three-months ended December 31, 2024. This increase primarily reflects higher non-cash stock-based compensation expenses in the current period, due to the timing of grants and the related vesting, as well as increased personnel and professional fees to support reporting requirements and expanded operations.
Other income decreased by approximately $819 to $402 for the three-months ended December 31, 2025, as compared to $1,221 for the three-months ended December 31, 2024. This decrease primarily reflects reduced foreign exchange impacts resulting from the change in the functional currency from Canadian dollars to US dollars, effective January 1, 2025.
During the three months ended December 31, 2025, the Company’s financial condition and cash flows reflected continued investment in clinical development activities. Cash and cash equivalents decreased during the quarter primarily due to operating cash outflows associated with increased research and development expenditures, including costs related to ongoing clinical studies and trial preparation activities. In addition, the Company continued to manage its liquidity by allocating funds between cash and cash equivalents and short-term investments, resulting in movements between these balances during the quarter. Increases in prepaid expenses related to clinical activities, along with the timing of accounts payable and accrued liabilities, also impacted cash during the period.
Liquidity and Capital Resources
Since inception, the Company has devoted its resources to funding R&D programs, including securing intellectual property rights and licenses, conducting discovery research, manufacturing drug supplies, conducting preclinical and clinical studies, and providing administrative support to R&D activities, which has resulted in an accumulated deficit of $81,970 as of December 31, 2025. With no current revenues, losses are expected to continue while the Company’s R&D programs are advanced.
We currently do not earn any revenues from our product candidates and are therefore considered to be in the development stage. As required, the Company will continue to finance its operations through the sale of equity or pursue non-dilutive funding sources available to the Company in the future. The continuation of our research and development activities for our muscle regeneration platform is dependent upon our ability to successfully finance and complete our research and development programs through a combination of equity financing and revenues from strategic partners. We have no current sources of revenues from strategic partners. Management has forecasted that the Company’s current level of cash will be sufficient to execute its current planned expenditures for the next 12 months without further financing.
Cash Management
At December 31, 2025, the Company had cash and cash equivalents and short-term investments of $27,710, compared with $48,548 of cash and cash equivalents and short-term investments at December 31, 2024. The decrease primarily reflects cash used to fund ongoing operations, particularly clinical trial costs and supporting operating activities. The Company invests cash in excess of operational requirements in highly rated and liquid investments.
Subsequent to the year end, on February 9, 2026, the Company completed a public offering of 5,168,019 Common Shares at US$10.10 per Common Share and 495,049 pre-funded warrants to purchase Common Shares at US$10.09999 per warrant (CA$13.80999 per warrant). Gross proceeds from the public offering were approximately $57,200.
Cash Flows:
The following table presents a summary of our cash flows for the years ended December 31, 2025, and 2024:
| | Year ended | | Year ended |
| | December 31, 2025 | | December 31, 2024 |
| | $ | | $ |
Net cash provided by/(used in): | |
| |
|
Operating activities | | (23,596) | | (18,252) |
Financing activities | | 2,241 | | 37,574 |
Investing activities | | (9,417) | | 4,656 |
Effect of foreign exchange on cash and cash equivalents | | 503 | | (586) |
Net (decrease)/increase in cash and cash equivalents | | (30,269) | | 23,392 |
18
Cash used in operating activities
Our cash used in operating activities for the year ended December 31, 2025, and 2024 was $23,596 and $18,252, respectively. Our uses of cash for operating activities consisted of costs related to the completion of the Phase 1, TRAILHEAD and BASECAMP clinical study, salaries and wages for our employees, fees paid in connection with preclinical studies, drug manufacturing costs, and professional fees.
Cash used in financing activities
Our cash flow from financing activities for the year ended December 31, 2025, consisted of proceeds from the exercise of warrants of $2,094, and proceeds from the exercise of stock options of $147. Our cash flow from financing activities for the year ended December 31, 2024, consisted of proceeds from the exercise of warrants of $600, net proceeds from share issuances from the December Equity Offering (as defined below) of $30,057, and net proceeds from pre-funded warrant issuances from the December Equity Offering of $6,917.
Cash used in investing activities
Our cash flow used in investing activities for the year ended December 31, 2025, was $9,417, and consisted of net purchases of investments of $9,411 and purchase of property and equipment of $6. Our cash flow from investing activities in the year ended December 31, 2024, was $4,656, and consisted of net maturity of investments of $4,660 and purchases of property and equipment of $4.
On February 9, 2026, the Company completed a public offering of 5,168,019 Common Shares at US$10.10 per Common Share and 495,049 pre-funded warrants to purchase Common Shares at US$10.09999 per warrant (CA$13.80999 per warrant). Gross proceeds from the public offering were approximately $57,200.
Satellos’ main objectives in managing capital are to ensure cash resources are preserved and provide sufficient liquidity to finance research and development activities, ongoing administrative costs and general operating requirements. Since inception, Satellos has financed its operations from private sales of equity, public sales of equity, convertible debt financing, non-convertible debenture financing, government grants and investment tax credits. Since Satellos has not generated net earnings from operations, its ongoing liquidity depends on its ability to access capital markets, which depends on the success of Satellos’ ongoing research and development programs, as well as capital market conditions.
The Company manages its capital structure in an endeavour to ensure sufficient resources are available to meet day-to-day operational requirements, further develop its existing technology, and continue as a going concern. In order to maintain or adjust the capital structure, the Company may issue new shares, issue debt or sell assets. Total capital is calculated as the Company’s own equity. The Company is not subject to any externally imposed capital requirements.
Satellos uses cash flow forecasts to estimate cash requirements and has forecasted that our existing cash and cash equivalents, and short-term investments, together with funds raised subsequent to the year ended December 31, 2025 is sufficient to operate the Company and meet our announced goals for the ensuing twelve months.
Based on future requirements, Satellos plans to raise capital as required to provide the necessary financial resources for operations. The timing of financings will depend on market conditions and Satellos’ cash requirements. Satellos’ cash flow forecasts are continually updated to reflect actual cash inflows and outflows to monitor the requirements and timing for additional financial resources. Satellos will continue to pursue various funding options and opportunities; however, no assurances can be made that Satellos will be successful
19
in raising additional investment capital, to continue as a going concern. Our ability to raise additional funds could be affected by adverse market conditions, the status of our product pipeline, and various other factors and we may be unable to raise capital when needed, or on terms favorable to us. If the necessary funds are not available, we may have to delay, reduce the scope of, or eliminate some of our development programs, potentially delaying the time to market for any of our product candidates.
Equity Offering December 2024 On January 27, 2026, the Company completed a twelve-for-one share consolidation of its issued and outstanding Common Shares. Any quantities relating to these instruments or any per unit price disclosed, have not been retrospectively adjusted for the share consolidation except for the weighted average number of shares outstanding used in the calculation of basic and diluted net loss per share and the Outstanding Share Data.
On December 20, 2024, the Company completed a public offering (the “December Equity Offering”), issuing 51,420,000 Common Shares at CA$0.90 per Common Share and 11,865,000 pre-funded warrants to purchase Common Shares with no expiry date and an exercise price of CA$0.00001 for CA$0.89999 per Pre-Funded Warrant for gross proceeds of $40,000.
The costs associated with the December Equity Offering were $3,100, including cash costs for commissions to the agents of approximately $2,781, professional fees and regulatory costs of $245, and accrued professional and regulatory fees of $74.
Bloom Burton Securities Inc. (“BBSI”), an entity jointly controlled by a director of Satellos, acted as exclusive agent and book running manager for the December Equity Offering.
USE OF PROCEEDS
December 2024 Financing
The following table provides an update on the milestones for the Duchenne program and the anticipated use of proceeds raised as part of the December Equity Offering (as previously proposed in the final prospectus dated December 17, 2024, relating to the December Equity Offering (the “December 2024 Prospectus”), along with the amounts actually expended.
| | Amount to Spend | | | | | |||
| | (as proposed in the | | | | | |||
| | December 2024 | | Costs Incurred | | Estimated | |||
Development Milestone | | Prospectus) | | to Date | | Remaining Costs | |||
BASECAMP clinical development of SAT-3247, including TRAILHEAD study and supporting CMC and pre-clinical activities | | $ | 28,092 | | $ | 11,192 | | $ | 16,900 |
General corporate and administrative expenses | | $ | 8,758 | | $ | 1,137 | | $ | 7,621 |
Total | | $ | 36,850 | | $ | 12,329 | | $ | 24,521 |
Please refer to the “Achievements and Highlights in the year ended December 31, 2025” section above for progress made during the period on the development milestone.
License Agreements
Ottawa Hospital Research Institute (“OHRI”)
Effective May 1, 2018, Satellos and OHRI entered into the OHRI License Agreement whereby OHRI granted Satellos an exclusive, world-wide, sublicensable, royalty bearing right and license to a body of technology and patents comprised of five patent families to develop, make, have made, import, use, offer for sale, sell and have sold or otherwise commercialize licensed products. At the same time the parties entered into a sponsored
20
research agreement, during the term of which OHRI has agreed to carry out specific research and development activities according to a prescribed statement of work, as may be amended from time to time, under the direction of the Company’s co-founder, Dr. Michael A. Rudnicki (the “OHRI SRA”). Under the OHRI SRA, Dr. Rudnicki leads a dedicated R&D team who are engaged solely to execute the agreed R&D program of Satellos, under his direction and as defined in the statement of work.
Long-Term Obligations and Other Contractual Commitments
The Company enters into contracts in the normal course of business, including for research and development activities. As at December 31, 2025, in addition to amounts that have been recognized in accounts payable and accrued liabilities, the Company has commitments for research and development activities in the amount of $23,142. These commitments are generally cancellable with notice, subject to payment for services rendered to the date of termination. These commitments include agreements related to the conduct of long-term toxicology, manufacturing, clinical development, and clinical trial costs.
| | Payments Due by Period | |||||||||||
| | Total | | Less than 1 year | | 1 -3 years | | 4 – 5 years | | After 5 years | |||
Purchase obligations | | $ | 23,142 | | $ | 14,686 | | $ | 8,456 |
| nil |
| nil |
The Company may be required to make annual, milestone, royalty, and other research and development funding payments to OHRI under the OHRI SRA and the OHRI License. These payments are contingent upon the achievement of specific development, regulatory and/or commercial milestones. The Company’s significant contingent milestone, royalty and other research and development commitments are as follows:
| ● | Royalties on net sales of any products covered by patents licensed from OHRI (“Licensed Products”) of 1% or 2% (depending on which patents cover a particular product), during the period when the applicable patents have valid, unexpired claims, subject to certain royalty stacking provisions; |
| ● | The following payments to OHRI may be triggered by specified events: |
o | CA$50 - each time a Licensed Product is the subject of an approved IND in the US or equivalent in any other industrialized country (maximum one payment per new drug candidate); |
o | CA$150 - each time a Licensed Product first enters Phase II human clinical trials in the US or equivalent in any other industrialized country (maximum one payment per new drug candidate); |
o | CA$300 - each time a Licensed Product first enters Phase III human clinical trials in the US or equivalent in any other industrialized country (maximum one payment per new drug candidate); and |
o | CA$1,000 - each time a Licensed Product is the subject of a regulatory approval in the US (such as NDA and BLA) or equivalent in any other industrialized country (maximum one payment per new drug candidate). |
| ● | 2% of sublicensing income received by Satellos from the grant of sublicenses. |
The Company has not accrued any amounts for these payments as of December 31, 2025, no milestones were achieved during the year. During the year ended December 31, 2024, the Company made a milestone payment of $37 upon the approval of an Investigational New Drug (IND) application for SAT-3247, in accordance with the agreement.
Subsequent to December 31, 2025, the Company made a milestone payment of $150 upon the initiation of the Phase 2 clinical trial for SAT-3247 in the US, in accordance with the agreement with OHRI.
21
TRANSACTIONS WITH RELATED PARTIES
The following related parties have engaged in transactions with the Company during the year ended December 31, 2024:
| a) | Bloom Burton Securities Inc. (“BBSI”) - an entity that is jointly controlled by Brian Bloom, a director of the Company. BBSI acted as lead agent in the December 2024 equity offering (Note 8). Related to the December Equity Offering on December 17, 2024, the Company paid $3,960 in commission and reimbursed BBSI for $141 in legal and related fees. |
| b) | Mr. William Jarosz, previously the Chief Executive Officer of iCo Therapeutics Inc. (“iCo”), the entity the Company completed a reverse takeover transaction with on August 13, 2021, and a former Director of the Company, had provided consulting services to iCo that were unpaid as of the date of the reverse takeover and this liability was assumed by the Company. Following the reverse takeover, Mr. Jarosz provided consulting services to the Company until November 2023. During the year ended December 31, 2024, the Company fully settled the outstanding balance of $706. |
Key management personnel consist of the Company’s Chief Executive Officer, Chief Scientific Officer, Chief Medical Officer, former Chief Business Officer, Chief Financial Officer and the Directors of the Company. The remuneration of key management personnel is as follows:
| | Year ended | | Year ended |
| | December 31, 2025 | | December 31, 2024 |
| | $ | | $ |
Salaries and management fees |
| 2,676 |
| 2,299 |
Stock-based compensation |
| 2,060 |
| 1,093 |
Total |
| 4,736 |
| 3,392 |
OFF-BALANCE SHEET ARRANGEMENTS
Satellos has not entered into any material off-balance sheet arrangements.
FINANCIAL INSTRUMENTS AND RISK MANAGEMENT
Satellos is exposed to various risks through its financial instruments as at December 31, 2025. The Company’s risk exposures and the impact on the Company’s financial instruments are summarized below:
Credit Risk
Credit risk arises from cash and cash equivalents and short-term investments held at banks and financial institutions, as well as outstanding receivables. The carrying value of these items represent the Company’s maximum exposure to credit risk. At December 31, 2025 and 2024, no expected credit losses were recognized on any outstanding receivables. During the year ended December 31, 2025, the Company invested its excess cash in interest-bearing operating accounts held at a Schedule 1 Canadian bank and in US government treasury bills and Guaranteed Investment Certificates. The Company limits its exposure to credit risk, with respect to cash and cash equivalents and short-term investments, by maintaining cash balances with large, reputable financial institutions and by investing in highly liquid instruments issued or guaranteed by governments or financial institutions. Such investments are restricted to instruments with a minimum credit rating of BB (or equivalent) at the time of investment. The Company’s cash equivalents and short-term investments consist primarily of operating funds, US government treasury bills, deposit investments and Guaranteed Investment Certificates with commercial banks. The carrying values of cash and cash equivalents, short-term investments, and receivables represent the Company’s maximum exposure to credit risk.
22
Liquidity Risk
Liquidity risk is the risk that the Company will encounter difficulty in raising funds to meet cash flow requirements associated with financial instruments. The Company controls liquidity risk through management of cash and cash equivalents, short-term investments, cash flows, and the availability and sourcing of financing. The Company’s ability to accomplish all of its future strategic plans is dependent on obtaining additional financing or executing other strategic options; however, there is no assurance the Company will achieve these objectives. Management monitors the Company’s liquidity position based on its existing cash and cash equivalents and short-term investments, together with expected cash requirements. As at December 31, 2025, the Company’s liabilities consist of accounts payable and accrued liabilities that have contracted maturities of less than one year.
Market Risk
| a) | Currency Risk |
Currency risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in foreign exchange rates. The exposure to this risk changes as the exchange rate fluctuates. Foreign currency risk is limited to the portion of the Company’s business transactions denominated in currencies other than the US dollar. The Company manages foreign exchange risk by maintaining Canadian dollars in cash on hand to fund its short-term foreign currency expenditures. Balances held in foreign currencies, presented in US dollars are as follows:
| | As at December 31, 2025 | ||||||||
| | US | | Australian | | Euro | | Canadian | | Total |
| | $ | | $ | | € | | $ | | $ |
Cash and cash equivalents | | 5,105 | | 498 | | — | | 4,201 | | 9,804 |
Short-term investments | | 14,550 | | — | | — | | 3,356 | | 17,906 |
Accounts payable and accrued liabilities |
| (1,759) |
| (4) |
| (921) |
| (1,421) |
| (4,105) |
Total |
| 17,896 |
| 494 |
| (921) |
| 6,136 |
| 23,605 |
| | As at December 31, 2024 | ||||||||
| | US | | Australian | | Euro | | Canadian | | Total |
| | $ | | $ | | € | | $ | | $ |
Cash and cash equivalents | | 24,848 | | 339 | | — | | 14,886 | | 40,073 |
Short-term investments |
| 5,000 |
| — |
| — |
| 3,475 |
| 8,475 |
Accounts payable and accrued liabilities |
| (2,491) |
| (329) |
| (242) |
| (521) |
| (3,583) |
Total |
| 27,357 |
| 10 |
| (242) |
| 17,840 |
| 44,965 |
Assuming all other variables remain constant, a 10% depreciation or appreciation of the US dollar against the Canadian dollar, Australian dollar, and Euro would result in an increase or decrease in loss and comprehensive loss for the year ended December 31, 2025, of $571 (December 31, 2024 - $1,761).
| b) | Interest Rate Risk |
Interest rate risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in market interest rates. The Company holds its cash and cash equivalents and short-term investments in banks and financial institutions and manages its interest rate risk by holding cash in high yield savings accounts or highly liquid short-term investments.
23
| c) | Fair Value |
Financial assets and liabilities are recognized on the statement of financial position at amortized cost in a hierarchy that is based on significance of the inputs used in making the measurements. The levels in the hierarchy are:
| ● | Level 1 – Quoted prices (unadjusted) in active markets for identical assets or liabilities |
| ● | Level 2 – Inputs other than quoted prices included within level 1 that are observable for the asset or liability, either directly (i.e., as prices) or indirectly (i.e., derived from prices) |
| ● | Level 3 – Inputs for the asset or liability that are not based on observable market data (i.e., unobservable inputs) |
At December 31, 2025, the Company’s financial instruments included cash and cash equivalents, short-term investments, and accounts payable and accrued liabilities.
Due to the short-term maturities of cash and cash equivalents, short-term investments and accounts payable and accrued liabilities, the carrying amounts approximate fair value at the respective consolidated statement of financial position date.
CRITICAL ACCOUNTING ESTIMATES
The preparation of consolidated financial statements in accordance with IFRS Accounting Standards requires management to make judgements, estimates, and assumptions that affect the application of accounting policies and reported amounts of assets, liabilities, revenues and expenses. Actual results may differ from these estimates. Estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimates are revised and in any future periods affected. Management has applied significant judgements, estimates and assumptions to the following:
a)Research and development expense estimates
The Company records research and development expenses based on estimates of services performed and costs incurred by contract research organizations, contract manufacturing organizations, clinical trial sites, and other third-party vendors. These estimates include accrued expenses and prepayments for research and development activities. As vendors may invoice in arrears or in advance, management estimates the costs incurred but not yet invoiced at each reporting date. Estimates are based on factors such as project progress, service reports, milestones, and contract terms. Due to the nature of these arrangements, significant judgment is required in determining the completeness of accruals and prepayments, and actual amounts may differ from management’s estimates.
b)Impairment of intangible assets
Impairment exists when the carrying value of an asset or cash generating units exceeds its recoverable amount, which is the higher of its fair value less costs of disposal and its value in use. During the year ended December 31, 2024, management determined that it was no longer likely that the sale of AmpB would be completed. The Company recognized an impairment of $2,905 to fully write down the remaining carrying value of the intangible asset. The factor leading to this impairment was the lack of financing obtained by NW Micelle Therapeutics Inc. to advance the technology. On October 7, 2024, the Company exercised its Put Option, and NW Pharmatech Limited did not fulfil their obligations under the terms of the agreement. On July 29, 2025, the Company completed the sale of AmpB, to Natural Works Trading Limited.
24
CHANGES IN ACCOUNTING POLICIES
Effective January 1, 2025, the Company changed its presentation currency from CA to USD. Refer to “Change in Presentation Currency” for further details.
DISCLOSURE CONTROLS AND INTERNAL CONTROL OVER FINANCIAL REPORTING
The Company has implemented a system of internal controls that it believes adequately protects the assets of the Company and is appropriate for the nature of its business and the size of its operations. The internal control system was designed to provide reasonable assurance that all transactions are accurately recorded, that transactions are recorded as necessary to permit preparation of financial statements in accordance with IFRS Accounting Standards and that our assets are safeguarded.
Internal control over financial reporting means a process designed by or under the supervision of the Chief Executive Officer and the Chief Financial Officer to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with IFRS Accounting Standards. The internal controls are not expected to prevent and detect all misstatements due to error or fraud.
These internal controls include disclosure controls and procedures designed to ensure that information required to be disclosed by the Company is accumulated and communicated as appropriate to allow timely decisions regarding required disclosure.
There were no changes in our internal control over financial reporting that occurred during the year ended December 31, 2025 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
As of December 31, 2025, the Company’s management assessed the effectiveness of our internal control over financial reporting and disclosure controls and procedures using the Committee of Sponsoring Organizations of the Treadway Commission’s 2013 framework. Based on their evaluation, the Chief Executive Officer and the Chief Financial Officer have concluded that these controls and procedures are effective.
OUTSTANDING SHARE DATA
As of the date of this MD&A, the Company had the following issued and outstanding securities:
Security | | Number |
Common shares | | 20,831,190 |
Pre-Funded Warrants | | 3,450,522 |
Stock options | | 2,194,453 |
On January 27, 2026, subsequent to the reporting period, the Company completed the Consolidation. The outstanding share data presented in this table have been retroactively adjusted to reflect the Consolidation.
25
RISKS AND UNCERTAINTIES
We are a development stage biopharmaceutical company that operates in an industry that is dependent on a number of factors that include the capacity to raise additional capital on reasonable terms, obtain positive results of clinical trials, obtain positive results of clinical trials without serious adverse or inappropriate side effects, and obtain market acceptance of its product. An investment in our Common Shares is subject to a number of risks and uncertainties. An investor should carefully consider the risks described in our AIF, as well as our other public filings with the securities regulators before investing in our Common Shares. If any of such described risks occur, or if others occur, our business, operating results and financial condition could be seriously harmed, and investors may lose a significant proportion of their investment. There are important risks which management believes could impact our business. For information on risks and uncertainties, please refer to the “Risk Factors” section of our most recent AIF filed on SEDAR+ at www.sedarplus.com.
ADDITIONAL INFORMATION
Additional information related to Satellos, including the AIF, is available by accessing the Company’s SEDAR+ profile at www.sedarplus.com.
26