Exhibit 99.1

SATELLOS BIOSCIENCE INC.
ANNUAL INFORMATION FORM
For the Year Ended
December 31, 2025
March 27, 2026
Exhibit 99.1
SATELLOS BIOSCIENCE INC.
ANNUAL INFORMATION FORM
For the Year Ended
December 31, 2025
March 27, 2026
3 | |
CAUTION REGARDING FORWARD-LOOKING STATEMENTS AND RISK FACTORS | 4 |
8 | |
9 | |
14 | |
28 | |
57 | |
58 | |
59 | |
60 | |
61 | |
68 | |
70 | |
71 | |
72 | |
73 | |
74 | |
75 | |
76 | |
A-1 |
In this annual information form (“Annual Information Form” or “AIF”), unless the context requires otherwise, references to the “Company”, “Satellos”, “we”, “us”, “our” and similar words refer to Satellos Bioscience Inc. or any predecessor thereto and all subsidiaries of the Company, as the context requires. All dollar amounts in this Annual Information Form are in Canadian dollars, except where otherwise indicated.
The information in this Annual Information Form is presented as of December 31, 2025, unless otherwise indicated.
All references to common shares in the capital of the Company (the “Common Shares”) herein refer to such Common Shares following the share consolidation which occurred on January 27, 2026 on the basis of one post-consolidation Common Share for every 12 pre-consolidation Common Shares (the “Consolidation”). Unless otherwise noted, such Common Shares are presented on a post-Consolidation basis.
MARKET AND INDUSTRY DATA
This AIF includes market and industry data that has been obtained from third party sources, including industry publications. The Company believes that its industry data is accurate and that its estimates and assumptions are reasonable, but there is no assurance as to the accuracy or completeness of this data. Third party sources generally state that the information contained therein has been obtained from sources believed to be reliable, but there is no assurance as to the accuracy or completeness of included information. Although the data is believed to be reliable, the Company has not independently verified any of the data from third party sources referred to in this AIF or ascertained the underlying economic assumptions relied upon by such sources.
- 3 -
CAUTION REGARDING FORWARD-LOOKING STATEMENTS AND RISK FACTORS
Certain statements and information in this Annual Information Form contain forward-looking statements or forward- looking information under applicable securities legislation that may not be based on historical fact, including, without limitation, statements containing the words “believe”, “may”, “plan”, “will”, “estimate”, “continue”, “anticipate”, “intend”, “expect”, “predict”, “project”, “potential”, “pursue”, “ongoing”, “could”, “would”, “seek”, “views”, “attempt”, “target”, “goal”, and “foresee” or any derivation or negative thereof or other comparable terminology, although not all forward-looking statements contain these words and similar expressions.
Forward-looking statements are necessarily based on estimates and assumptions made by us in light of our experience and perception of historical trends, current conditions and expected future developments, as well as factors that we believe are appropriate. Forward-looking statements in this Annual Information Form include, but are not limited to, statements relating to:
| ● | our belief that the Company will be successful in raising additional capital to continue as a going concern; |
| ● | the expected research and development timelines, therapeutic benefits, effectiveness and safety of our product candidates; |
| ● | our belief that the Company’s products and research and development efforts are targeting diseases and conditions with significant unmet medical treatment needs; |
| ● | our belief that the Company has made, and will continue to make progress towards the achievement of certain milestones or objectives; |
| ● | our expectation with respect to meeting milestones and the minimum amount of funds the Company expects to need to raise in order to achieve such milestones and garner additional funding; |
| ● | the initiation, timing, cost, progress, outcomes, resource needs and success of our research and development activities, plans and programs; |
| ● | our expectations regarding our ability to design, test and patent novel drug products suitable for advancement into clinical trials and the anticipated timelines surrounding such clinical trials; |
| ● | our belief that we will not receive substantive comments on our Investigational New Drug (“IND”) or equivalent applications; |
| ● | our expectations that the Notch pathway and AAK1 drug target (both as further described herein) represent drug development opportunities similar or superior to modulation of the EGFR signaling pathway; |
| ● | our intentions of developing inhibitors to AAK1 (including but not limited to SAT-3247 and SAT-3153) and in showing that such potential inhibitors have desirable effects in relevant models of Duchenne muscular dystrophy (“Duchenne” or “DMD”) and in other indications, such as other degenerative muscle diseases, muscle injury or trauma, or muscle regeneration generally; |
| ● | our expectations that we will identify predictive biomarkers which will translate into or be useful in conducting human clinical trials; |
| ● | our belief that the results of Satellos’ research and development activities, preclinical studies, safety studies or clinical trials have the potential to be commercially competitive with research and development activities, preclinical studies, safety studies or clinical trials conducted by other parties; |
| ● | discoveries we have made in muscle stem cell regulation having the potential to represent insights into a potential root cause of degenerative muscle disorders which has previously not been recognized and which may be therapeutically relevant in the treatment of degenerative muscle disorders; |
| ● | our belief that the Company’s technology can be commercialized, and that such commercialization could be done as effectively or more effectively than other technologies to treat degenerative muscle disorders and conditions or other medical disorders or conditions, or at all; |
| ● | our ability to discover, optimize, select and advance into clinical development therapeutic drug development candidates in a timely, cost-efficient and effective manner, or at all; |
| ● | our ability to translate our discoveries in muscle stem cell regulation into safe and therapeutically effective drug products and the broad applicability of such products; |
| ● | our ability to enter into research and/or commercial development collaborations or partnerships to successfully and profitably advance our drug development candidates; |
| ● | our ability and that of our partners (if any) to advance identified drug development candidates into, and successfully complete, clinical trials; |
| ● | our intention to identify and nominate one or more back-up drug candidates and the potential benefits of having such back-ups; |
| ● | our plans to utilize and deploy MyoRegenXTM in our programs and our continued relationship with the Ottawa Hospital Research Institute (“OHRI”); |
- 4 -
| ● | our ability to develop the Company’s novel discoveries into viable therapeutic treatments suitable for clinical development, including, but not limited to, our ability to determine appropriate dosing regimens; |
| ● | the ability of our products to effectively and safely treat Duchenne and other degenerative muscle disorders and conditions or other medical disorders or conditions and the applicability of our products to other disorders and conditions; |
| ● | our expectations regarding future enrolment into clinical trials and the timing of future enrolment into clinical trials for our product candidates; |
| ● | our belief that our approach may reduce the risk, time and cost of developing therapeutics by avoiding some of the uncertainty associated with certain research and preclinical stages of drug development; |
| ● | our ability to establish and maintain relationships with collaborators with acceptable preclinical and/or clinical research and development capability and regulatory and commercialization expertise to enable the development and future commercialization of our technology or products and the benefits to be derived from such collaborative efforts; |
| ● | our ability to enter into agreements or partnerships with pharmaceutical or biotechnology companies that have research and clinical development and/or sales and marketing capabilities and the expected benefits that could be derived therefrom; |
| ● | our ability to generate and protect our intellectual property; |
| ● | our ability to operate our business without infringing upon the intellectual property rights of others; |
| ● | our ability to engage third party services with specialized domain expertise for the drafting and submitting of regulatory applications to conduct clinical trials in humans; |
| ● | our ability to establish suitable chemistry, manufacturing and controls and Good manufacturing procedures protocols; |
| ● | the manufacturing capacity of third-party manufacturers for our product candidates; |
| ● | our expectations regarding federal, provincial and foreign regulatory requirements; |
| ● | the timing of, and the costs of obtaining and maintaining, regulatory approvals in the United States, Australia, Europe, Serbia and other jurisdictions; |
| ● | the rate and degree of market acceptance and clinical utility of our future products, if any; |
| ● | existing and future corporate alliances and licensing transactions with third parties, and the receipt and timing of any payments to be made by us or to us pursuant to such arrangements; |
| ● | the implementation and execution of our commercial and operational strategy; |
| ● | our ability to engage and retain the consultants or employees required to grow our business; |
| ● | the potential revenue that may be generated from our products, pricing and reimbursement of the patient cost of our drug products by insurers or national health systems, as the case may be, in those jurisdictions where the Company intends to sell its drug products and our ability to achieve profitability; |
| ● | developments relating to our competitors and our industry, including the success of competing therapies that are or become available; |
| ● | the potential growth of the market and demand for our products as well as the estimated pricing and subsequent revenue generation of any potential therapeutics we discover; |
| ● | our future financial performance, including projected expenditures, future revenue, capital requirements and our needs for additional financing; |
| ● | our belief that our current liquidity position is sufficient to finance our operations through 2027; |
| ● | our intention to initiate a Phase 2 clinical trial in FSHD and the anticipated timing of regulatory filings therefor; |
| ● | our expectations regarding the conduct and outcomes of our ongoing BASECAMP and TRAILHEAD clinical studies and our plans to expand these studies to additional clinical sites and jurisdictions; |
| ● | our ability to maintain our listings on the TSX and Nasdaq and the continued liquidity and trading market for our Common Shares; |
| ● | the potential impact of tariffs, trade policy changes, and geopolitical conditions on our operations and ability to conduct business; |
| ● | general business and economic conditions and the evolving regulatory landscape; and |
| ● | regulatory and legislative changes that may result from geopolitical events or changes in government administration. |
- 5 -
Such forward-looking statements reflect our current views with respect to future events, are subject to risks and uncertainties and are necessarily based upon a number of estimates and assumptions that, while considered reasonable by Satellos as of the date of such statements, are inherently subject to significant medical, scientific, business, economic, competitive, political and social uncertainties and contingencies. Many factors could cause our actual results, performance, achievements, prospects or opportunities to be materially different from any future results, performance or achievements that may be expressed or implied by such forward-looking statements. In making the forward-looking statements included in this Annual Information Form, the Company has made various material assumptions, including, but not limited to:
| ● | obtaining positive results from our research and development activities, including clinical trials; |
| ● | our ability to obtain regulatory approvals; |
| ● | assumptions regarding general business, market, economic and regulatory conditions; |
| ● | assumptions regarding the cost and timing of each study; |
| ● | the Company’s ability to successfully advance its preclinical and clinical development programs and execute its plans substantially as currently envisioned; |
| ● | the Company’s ability to identify and advance suitable drug candidates; |
| ● | assumptions related to the pricing and reimbursement of its drug products in jurisdictions in which the Company intends to sell its drug products; |
| ● | the Company’s current positive relationships with third parties will be maintained and the potential to develop new partnerships; |
| ● | our ability to continue to use existing licenses for the development of our product(s); |
| ● | the availability (and sources) of financing on reasonable terms; |
| ● | future expenditures to be incurred by the Company, including research and development and operating costs; |
| ● | the Company’s ability to attract and retain skilled consultants and employees; |
| ● | assumptions regarding market competition, market capture and pricing; |
| ● | the products and technology offered by the Company’s competitors; and |
| ● | the Company’s ability to protect patents and proprietary rights. |
In evaluating forward-looking statements, current and prospective shareholders should specifically consider various factors, including the risks outlined herein under the heading “Risk Factors”. Certain risks and uncertainties that could cause such actual events or results expressed or implied by such forward-looking statements and information to differ materially from any future events or results expressed or implied by such statements and information include, but are not limited to:
| ● | risks related to the early stage of our products; |
| ● | uncertainties related to preclinical product development activities and clinical trial outcomes; |
| ● | uncertainties related to current economic conditions; |
| ● | risks related to rapid technological change; |
| ● | uncertainties related to forecasts and timing of clinical trials and regulatory approval; |
| ● | competition in the market for therapeutic products, including those to treat Duchenne and related diseases; |
| ● | risks related to potential product liability claims; |
| ● | availability of financing and access to capital and the risks associated with the Company’s ability to continue as a going concern; |
| ● | market acceptance and commercialization of products; |
| ● | the availability, costs and supply of materials; |
| ● | risks related to the effective management of our growth; |
| ● | risks related to the reliance on partnerships and licensing agreements; |
| ● | risks related to our reliance on key personnel; |
| ● | risks related to the regulatory approval process for the manufacture and sale of therapeutic products; |
| ● | risks related to the reimbursement process in various jurisdictions where the Company plans to sell its drug products; and |
| ● | our ability to secure and protect our intellectual property. |
- 6 -
The Company cautions that the foregoing list of important factors and assumptions is not exhaustive. Although the Company has attempted to identify on a reasonable basis important factors and assumptions related to forward-looking statements, there can be no assurance that forward-looking statements will prove to be accurate, as events or circumstances or other factors could cause actual results to differ materially from those estimated or projected and expressed in, or implied by, these forward-looking statements. Should one or more of these risks or uncertainties, or a risk that is not currently known to us, materialize, or should assumptions underlying those forward-looking statements prove incorrect, actual results may vary materially from those described herein. The forward-looking statements made herein are made as of the date of this Annual Information Form and we do not intend, and do not assume any obligation, to update these forward-looking statements except as required by applicable securities laws. Readers are cautioned that forward-looking statements are not guarantees of future performance and are inherently uncertain. Accordingly, readers should not put undue reliance on forward-looking statements.
- 7 -
General
Satellos Bioscience Inc. was incorporated under the Canada Business Corporations Act (the “CBCA”) on July 27, 2012 (“Pre-Arrangement Satellos”). iCo Therapeutics Inc. (“iCo”) was incorporated under the Business Corporations Act (British Columbia) on April 20, 2006 and completed a reverse take-over transaction by way of statutory arrangement onto the TSX Venture Exchange.
On August 13, 2021, Pre-Arrangement Satellos and iCo completed a plan of arrangement (the “Arrangement”) under section 192 of the CBCA, pursuant to which, among other things, iCo acquired all of the issued and outstanding shares of Pre-Arrangement Satellos. In connection with the Arrangement, iCo (now, Satellos): (a) was continued under the CBCA; (b) amalgamated with Satellos to form the Company as it now exists, which continues to carry on the pre-Arrangement business of Pre-Arrangement Satellos and iCo; and (c) consolidated its outstanding Common Shares on a 20:1 basis.
On February 14, 2024, the Company commenced trading on the Toronto Stock Exchange (the “TSX”) under the symbol “MSCL”, and was delisted from the TSX Venture Exchange effective as of the close of the market on February 14, 2024.
On January 27, 2026, the Company completed the Consolidation of the Common Shares on the basis of one post-Consolidation Common Share for every 12 pre-Consolidation Common Shares. The Consolidation took effect on the TSX at market open on January 30, 2026, and the Company commenced trading on the Nasdaq Global Market (“Nasdaq”) under the trading symbol “MSLE” on February 6, 2026.
The Company has two wholly owned subsidiaries, Satellos Bioscience Australia Pty Ltd. (an entity incorporated under the laws of Australia) and Satellos Bioscience US, Inc. (incorporated under the laws of Delaware, USA).
The Company’s head office is located at Royal Bank Plaza, South Tower, 200 Bay St., Suite 2800, Toronto, Ontario, M5J 2J1, and the Company’s registered and records office is located at Royal Bank Plaza, South Tower, 200 Bay St., Suite 2800, Toronto, Ontario, M5J 2J1.
Intercorporate Relationships
The following is a current corporate organizational chart of the Company:
| Notes: (1)The Company is the registered owner of 100% of the issued and outstanding shares of Satellos Bioscience US, Inc. (2)The Company is the registered owner of 100% of the issued & outstanding shares of Satellos Bioscience Australia Pty Ltd. |
- 8 -
GENERAL DEVELOPMENT OF THE BUSINESS
Three-Year History
A general description of the Company’s products and services is included below in “Description of the Business – General”. Below is a description of the relevant history of the Company and its predecessor entities over the last three completed financial years:
2023
On January 3, 2023, Satellos nominated SAT-3153 as its then pre-IND lead development candidate.
On February 6, 2023, the Company announced results from preclinical studies in the Mdx mouse model of Duchenne (the most common mouse model for Duchenne) indicating positive effects of modulating stem cell polarity with drug on muscle size and function. On March 9, 2023, the Company announced preclinical results using SAT-3153 in the Mdx mouse model of Duchenne indicating positive effects on stem cell polarity and muscle function, as well as an update on certain drug-like characteristics.
On March 24, 2023, the Company closed a debenture unit offering, pursuant to which the Company issued 2,385 debenture units and raised gross proceeds Cdn$2,385,000. The Company also issued 55,985 Common Shares as part of the units issued in the debenture unit offering. On August 14, 2023, the Company exercised its option to repay the Debentures.
On May 17, 2023, the Company closed the “May 2023 Equity Offering”, which offered Common Shares at Cdn$6.00 per Common Share and pre-funded Common Share purchase warrants (“Pre-Funded Warrants”) with no expiry date for Cdn$5.99988 per Pre-Funded Warrant. Investors purchased 5,858,101 Common Shares and 3,308,564 Pre-Funded Warrants for gross proceeds of Cdn$55 million. Each Pre-Funded Warrant is exercisable for one Common Share at an exercise price of Cdn$0.00012 per share. 615,326 compensation warrants were granted to the agents with each such compensation warrant was exercisable into one Common Share at an exercise price of Cdn$6.00 until expiry on May 17, 2025.
On June 7, 2023, Satellos announced the appointment of Alan K. Jacobs, M.D., as Chief Medical Officer of the Company.
On August 2, 2023, Satellos announced that the FDA granted Orphan Drug Designation and Rare Pediatric Disease Designation to SAT-3153 for the potential treatment of Duchenne muscular dystrophy.
On September 5, 2023, Satellos announced the appointment of Elizabeth Williams, CPA, CA as Chief Financial Officer (“CFO”) of the Company. Ms. Williams has extensive experience in biotech, working with publicly listed entities in both Canada and the United States.
On November 14, 2023, the Company disclosed for the first time that the drug target for the Duchenne program is AAK1 (formerly K9), a protein kinase in the Notch pathway, which the Company discovered can be modulated to enable muscle regeneration.
Throughout 2023 the Company continued its drug development efforts. As part of its ongoing efforts to advance SAT-3153 into clinical development, the Company generated additional data and made new observations informing the properties and characteristics for the purpose of developing an inhibitor of the protein AAK1. Through these preclinical efforts, the Company identified a new molecule, SAT-3247, and generated data which demonstrated that SAT-3153 and SAT-3247 have a similar capacity to affect muscle regeneration and functional benefit in the Mdx mouse model of Duchenne. SAT-3247 also exhibited improved oral bioavailability, target specificity and tissue distribution when compared directly to SAT-3153 in preclinical studies.
On November 14, 2023, Satellos announced that SAT-3247 was being nominated as the Company’s lead clinical development candidate with SAT-3153 becoming the back-up development candidate. Satellos then began conducting IND-enabling studies with SAT-3247. Satellos also announced the appointment of Ms. Courtney Wells as Senior Vice President of Clinical Development Operations.
2024
On January 23, 2024, the Company announced the departure of Alan Jacobs, M.D., as Chief Medical Officer and the appointment of Jordan Dubow, M.D. as Chief Medical Advisor.
On February 13, 2024, the Company announced positive preliminary data showing SAT-3247 can improve skeletal muscle function in a mouse model of facioscapulohumeral muscular dystrophy (“FSHD”). FSHD, an adult-onset muscular dystrophy that results in the
- 9 -
progressive destruction of muscle tissue, is the third most common muscular dystrophy behind Duchenne (& the related Becker muscular dystrophy) and myotonic dystrophy. In research conducted under a grant from the FSHD Canada Foundation, Satellos demonstrated that treatment with SAT-3247 successfully improved the disease phenotype FLExDUX4 mouse model of FSHD.
On February 14, 2024, the Company announced that it would commence trading on the TSX on February 15, 2024 under the symbol “MSCL”, and delist from the TSXV effective as of the close of the market on February 14, 2024.
On March 4, 2024, Satellos announced positive preclinical data presented at the Muscular Dystrophy Association Clinical and Scientific Conference showing that SAT-3247 improved skeletal muscle function in three mouse models of muscle degeneration, these models being: the mdx model, a gold standard research model bearing the same genetic defect as patients with Duchenne; the FLExDUX4 model of FSHD; and a muscle injury model in wildtype mice. In all instances, treatment with SAT-3247 over a three-to-four-week period resulted in a statistically significant improvement in muscle force versus animals receiving placebo showing the potential for broad clinical applicability of SAT-3247 in degenerative muscle conditions
On May 28, 2024, Satellos announced the formation of a Clinical Advisory Board comprised of distinguished physicians with expertise in DMD to support the advancement of SAT-3247 in clinical trial development.
On July 2, 2024, Satellos announced data in a canine model of DMD showing improved muscle repair and regeneration and improved muscle force from SAT-3247 treatment. After treatment with SAT-3247 the animals showed an increase in Regenerative Index, a measure of the number of newly regenerated muscle fibers versus the number of damaged and dying muscle fibers, suggesting that muscle repair and regeneration is occurring. These results were further updated on August 12, 2024 and October 1, 2024.
On July 11, 2024, the Company announced submission of a clinical research proposal to a Human Research Ethics Committee (HREC) in Australia seeking regulatory authorization under their Therapeutic Goods Administration’s (TGA’s) Clinical Trial Notification (CTN) scheme to conduct a first-in-human Phase 1 clinical trial of SAT-3247. Satellos announced on August 19, 2024 that the HREC submission had been approved.
On August 7, 2024, Satellos announced that the U.S. Food and Drug Administration (“FDA”) had granted Rare Pediatric Disease Designation to SAT-3247 for the potential treatment of DMD after receiving Orphan Drug Designation earlier in the year.
On September 19, 2024, Satellos announced that the first participant in the Company’s first-in-human Phase 1 clinical trial had been dosed. The Phase 1 clinical trial comprised two components. In the first component, Phase 1a, 72 healthy volunteers were enrolled in a blinded, randomized, placebo-controlled, staggered, parallel design study to assess the safety and pharmacokinetic properties of SAT-3247. Participants were randomized across five single-ascending dose (“SAD”) cohorts, four multiple-ascending dose (“MAD”) cohorts, and one food effect dose cohort. In the second component, Phase 1b, 5 adult volunteers with genetically confirmed DMD, ages 20 – 27, were enrolled in a 28-day, open-label, single dose cohort to assess safety and pharmacokinetic (“PK”) properties in patients and to explore potential pharmacodynamic markers. The study had the potential to enroll up to 10 adults.
On October 1, 2024, Satellos announced data to be presented at the 29th Annual Congress of the World Muscle Society which took place October 8-12, 2024, in Prague. The presentation provided an overview of key data collected during the open-label pilot study of SAT-3247 in a canine model of DMD. The data presented from the pilot study demonstrated that treatment of DMD canines with SAT-3247 improved measures of strength to near normal levels.
On November 14, 2024, the Company announced the appointment of Stephanie Brown to its Board of Directors. Ms. Brown brings over 30 years of biopharma industry experience, having held numerous executive roles contributing to achievements in product commercialization and organizational transformation.
On December 11, 2024, the Company announced the first adult participant with DMD had been dosed in the Phase 1b clinical trial.
On December 20, 2024, Satellos announced that it closed an equity offering, issuing a total of 5,273,750 equity securities for gross proceeds of approximately Cdn$57 million (USD$40 million) (the “December 2024 Public Offering”).
- 10 -
2025
Effective January 1, 2025, the Company changed its functional and presentation currency from the Canadian dollar to the United States dollar to better reflect the primary economic environment in which the Company currently operates, with the majority of future research and development activities expected to be USD-denominated.
On February 10, 2025, the Company announced that the five SAD cohorts, four MAD cohorts, and one cross-over food effect single-dose cohort of the Company’s Phase 1a clinical trial with SAT-3247 had been fully enrolled.
On March 19, 2025, the Company announced initial safety PK data of SAT-3247 from the Phase 1a clinical trial in an oral presentation at the 2025 Muscular Dystrophy Association Clinical & Scientific Conference.
In Phase 1a, 72 healthy volunteers were randomized across five SAD cohorts (including one cross-over food effect cohort) with single oral doses of up to 400 mg, and four MAD cohorts with daily oral doses up to 240 mg/day for 7 consecutive days. Phase 1a data showed that:
| ● | SAT-3247 was safe and well tolerated across all healthy volunteer cohorts. At predicted human efficacious dose levels (i.e., between 50 and 150 mg total daily dose), SAT-3247 did not display adverse clinical findings on any parameter measured, including clinical labs, vital signs, ECG’s, and physical exams. No moderate or greater drug-related adverse events were reported at any dose studied and any mild events reported were reversible. |
| ● | Phase 1a PK data demonstrated consistency with results from the Company’s preclinical studies. These PK results confirmed post-dose plasma concentrations of SAT-3247 were sustained at levels and time courses which the Company’s research findings suggest are most likely to yield a therapeutic effect on muscle regeneration and strength. |
On May 22, 2025, the Company announced data from the Phase 1b clinical study with SAT-3247.
On June 18, 2025, the Company announced the election of Iris Loew-Friedrich, M.D., Ph.D., and Selwyn Ho, MBBS, to its Board of Directors. As part of the Annual General Meeting, Rima Al-awar, Ph.D., William Jarosz, J.D., and William McVicar, Ph.D., did not stand for re-election.
On July 16, 2025, the Company announced the appointment of Wildon Farwell, M.D., MPH, as Chief Medical Officer. Dr. Farwell joined Satellos from Dyne Therapeutics (Nasdaq: DYN), where he served as Chief Medical Officer and medical advisor. At Dyne, Dr. Farwell built the development organization, led the protocol development and regulatory submissions for their DMD and myotonic dystrophy type 1 (DM1) programs, oversaw the conduct of multiple potentially registrational clinical studies, and contributed to several successful capital raises. Before joining Dyne, he spent a decade at Biogen in increasing leadership roles, including on clinical programs resulting in the approval of Spinraza and Qalsody as then-novel medicines for the treatment of the neuromuscular diseases SMA and ALS, respectively. Prior to moving into industry, Dr. Farwell served as an assistant professor of medicine at Harvard Medical School and was a physician at Brigham and Women’s Hospital and the VA Boston Healthcare System. He earned his medical degree from the University of Missouri School of Medicine and holds a Master of Public Health in clinical effectiveness from the Harvard T.H. Chan School of Public Health.
The Company announced on September 22, 2025, that 144,791 warrants with an exercise price of Cdn$7.20 had been exercised for gross proceeds of Cdn$1.0 million.
On September 22, 2025, the Company announced the submission of an IND application to the U.S. FDA, along with parallel regulatory filings in the United Kingdom, Europe, Serbia and Australia, to initiate a Phase 2 clinical trial of SAT-3247 in ambulatory children with DMD.
On October 10, 2025, at the 30th Annual Congress of the World Muscle Society in Vienna, Austria, the Company announced new clinical trial data from the Company’s Phase 1b clinical trial of SAT-3247 further demonstrating potential tolerability and providing a preliminary indication of potential for therapeutic efficacy of SAT-3247 following treatment over a period of 28 days, as follows:
| ● | Individuals treated with SAT-3247 over a 28-day period demonstrated an increase in grip strength far greater than seen in the Duchenne natural history in this age group. Specifically, a 118.6% mean improvement in maximum grip strength was observed |
- 11 -
| in the dominant hand and 97.9% mean improvement in the non-dominant hand, representing an approximate doubling of grip strength from ~2 kg to ~4 kg. These improvements are inconsistent with published natural history and were correlated with higher drug concentrations on Day 15 and higher baseline creatinine (a surrogate for increased muscle mass), which we believe indicates our drug is having the desired impact on muscle. The increases in grip strength seen in 3 of the 5 patients after 28 days appear to be moving into strength measures not seen in the natural history for patients in this age demographic. Improvements of this nature (if sustained) may provide an opportunity for early approval consideration of SAT-3247. |
| ● | Furthermore, participants exhibited a 5.8% mean improvement of predicted forced vital capacity; an increase that is also inconsistent with natural history with declines about 5% annually among adults with Duchenne. All other measures remained stable over the study period. No drug-related adverse events of moderate severity or higher were observed in either study, and no dose-limiting toxicities occurred. |
| ● | Data presented at the meeting also demonstrated that SAT-3247 was safe and well-tolerated with a desirable pharmacokinetic (PK) profile across the Phase 1a portion of the study conducted in 72 healthy adult human volunteers. |
Study participants from the Phase 1b were offered the option, upon completing the Phase 1b study, to enroll into an 11-month, long-term follow-up study of treatment with SAT-3247, known as LT-001 (“TRAILHEAD”).
On October 21, 2025, the Company announced that the first patient had been dosed in TRAILHEAD (aka LT-001).
On November 14, 2025, the Company announced that it had appointed Mark Nawacki CPA, CA, co-founder, president and former CEO of Searchlight Pharma, to its board of directors.
On December 9, 2025, the Company announced that it received IND clearance by the United States FDA, as well as other global regulatory agencies, to conduct SAT-3247-CL-201 (“BASECAMP”), a three-month, randomized, double-blind, placebo-controlled, proof-of-concept, Phase 2 study of SAT-3247 in 51 ambulatory children with DMD. In addition to the FDA clearance, the United Kingdom’s Medicine and Healthcare products Regulatory Agency granted authorization of the Company’s Clinical Trial Application (“CTA”); Australia’s Human Research Ethics Committee accepted the Therapeutic Goods Administration’s Clinical Trial Notification scheme for regulatory authorization; and the Medicines and Medical Devices Agency of Serbia approved the CTA. On December 12, 2025, the CTA was approved in Canada and on January 22, 2026, the CTA was approved by the European Union. The Company is currently cleared to proceed with 60mg and 120mg doses of SAT-3247 in the UK, Australia, Serbia and the European Union. The FDA and Health Canada cleared the Company to proceed with the 60mg dose but require the Company to provide additional pharmacokinetic data in pediatric patients before being cleared to proceed with the 120mg dose.
Subsequent Events
On January 27, 2026, the Company completed the Consolidation of its Common Shares on the basis of one post-Consolidation Common Share for every 12 pre-Consolidation Common Shares. The Consolidation took effect on the TSX at market open on January 30, 2026.
On January 29, 2026, the Company announced the appointment of Antoinette Paone as Chief Development Officer and Head of Regulatory Affairs. Ms. Paone brings extensive experience leading regulatory strategy from clinical development through approval, including over a decade at Vertex Pharmaceuticals (Nasdaq: VRTX) where as Vice President – North American Regulatory Strategy she contributed to the attainment of breakthrough status and FDA approvals of several novel small molecule drugs for cystic fibrosis including Kalydeco and Orkambi. She joins Satellos most recently from Generation Bio (Nasdaq: GBIO), where she served as Chief Operating Officer where she was instrumental in all aspects of the company’s growth from start-up to development ready organization.
On February 9, 2026, the Company announced that it had completed an underwritten public offering of 5,168,019 Common Shares, which included the exercise of the underwriters’ option to purchase an additional 712,574 Common Shares and, in lieu of Common Shares for certain investors, Pre-Funded Warrants to purchase 495,049 Common Shares (the “2026 Offering”). The Common Shares were sold at a price of US$10.10 per share (Cdn$13.81 per Common Share) and the Pre-Funded Warrants were sold at a price of US$10.09999 per Pre-Funded Warrant (Cdn$13.80999 per Pre-Funded Warrant), which represents the per share price for the Common Shares less the Cdn$0.00001 per share exercise price for each Pre-Funded Warrant. The total gross proceeds to the Company were approximately US$57.2 million, before deducting the underwriting discounts and commissions.
On February 12, 2026, the Company announced that the first participant had been dosed in BASECAMP.
- 12 -
On March 10, 2026, the Company announced further clinical and biomarker data for SAT-3247 at the Muscular Dystrophy Association (“MDA”) Clinical & Scientific Conference. The data included: results from a serum proteomic analysis from the previously completed 28-day, Phase 1b trial; interim functional data following a further 56 days of treatment in TRAILHEAD (nb., representing 84 days cumulatively); and the development of the Regenerative Index (“RI”) a novel muscle regeneration assessment tool to be used in assessing the impact of SAT-3247 treatment on muscle morphology in the BASECAMP trial. The Company also presented furhter preclinical findings demonstrating enhanced muscle strength in a mouse model of FSHD.
Significant Acquisitions
The Company did not complete any significant acquisitions (within the meaning of National Instrument 51-102 – Continuous Disclosure Obligations) in the financial year ended December 31, 2025.
- 13 -
The Company’s primary goal is the development of disease modifying therapeutic drugs for the treatment of severe muscle conditions of unmet medical need. Our core technology is based on discoveries by the Company’s scientific founder and Chief Discovery Officer, Dr. Michael Rudnicki, in understanding and modulating muscle stem cell function and its role in muscle regeneration. Multiple peer reviewed publications from Dr. Rudnicki’s lab (the “Rudnicki Lab”) at the OHRI have advanced the understanding of the identity and behavior of muscle stem cells including their role in health and disease. For instance, the Rudnicki Lab was the first to define so called muscle stem cells (a.k.a. ‘satellite stem cells’) and characterize a sub-population as bona fide multipotent stem cells capable of both self-renewal and regeneration (Source: Kuang et al., 2007, Cell). Dr. Rudnicki was also first to demonstrate that such stem cells exist as a special body of cells capable of regeneration, and subsequently elucidate their biological mechanism of action and identify means to modulate their activity. He further linked deficiencies in muscle stem cell function directly to the pathology of Duchenne as a potential causal factor in the progressive muscle destruction that occurs in this lethal disease (Source: Dumont et al., 2015, Nature Medicine).
The basic principle governing how muscle stem cells contribute to the creation of new muscle cells and hence muscle regeneration, through a process known as asymmetric division, is depicted below in Figure 1.
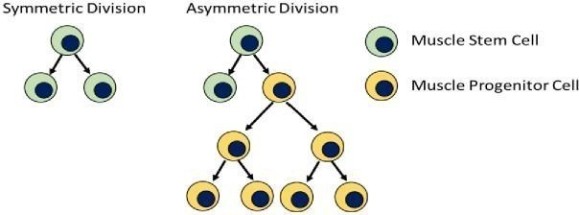
Figure 1: Muscle stem cells undergo symmetric or asymmetric divisions in response to injury stimuli. Muscle progenitor cells are generated to produce new muscle tissue or repair injured muscle.
Asymmetric muscle stem cell divisions result in one stem cell being produced and one progenitor muscle cell. The former maintains the pool of stem cells to be called on to respond to future injury. Progenitor muscle cells by contrast, continue to replicate through normal cell mitosis to generate potentially thousands of cells that ultimately incorporate into and become functional muscle tissue. Findings from the research of Dr. Rudnicki have linked deficits asymmetric division to the progressive muscle loss which is a principal pathology of DMD and other degenerative diseases.
To apply our understanding of muscle regeneration to therapeutic development in degenerative muscle conditions or disorders, Satellos employs a proprietary discovery platform developed by the Rudnicki laboratory at OHRI called, MyoReGenX™. An automated microscopy system, MyoReGenX™ recapitulates the muscle stem cell environment ex-vivo (i.e., outside the body) and enables Satellos to identify and assess opportunities for developing novel therapeutic treatments.
Lead Development Program: Duchenne
The Company’s first application of its technology is directed towards the discovery and development of a small molecule drug for the treatment of Duchenne, the most common fatal genetic disorder diagnosed in childhood affecting approximately one in 4,000 male births per year, worldwide. As depicted in the below Figure 2, individuals living with Duchenne experience severe and progressive loss of muscle function during their lives, often exhibited by loss of ambulation before their teenage years and generally culminating in death before the end of their third decade of life. There is no known cure.
Despite this dire scenario, Satellos takes hope for its novel approach from the fact that individuals living with Duchenne do make functional muscle as young children, albeit not as effective as their healthy peers. Our interpretation of the progressive nature of Duchenne, also depicted in Figure 2, is that the unmistakable signs of motor impairment and ambulatory challenges that become apparent during childhood represent a ‘tipping point’ in the balance between muscle damage and repair where regeneration fails to keep up with damage.
- 14 -
Satellos has designed SAT-3247 with the goal of resetting the balance of regeneration over degeneration by enhancing the process of asymmetric division and the ensuing creation of new muscle cells.
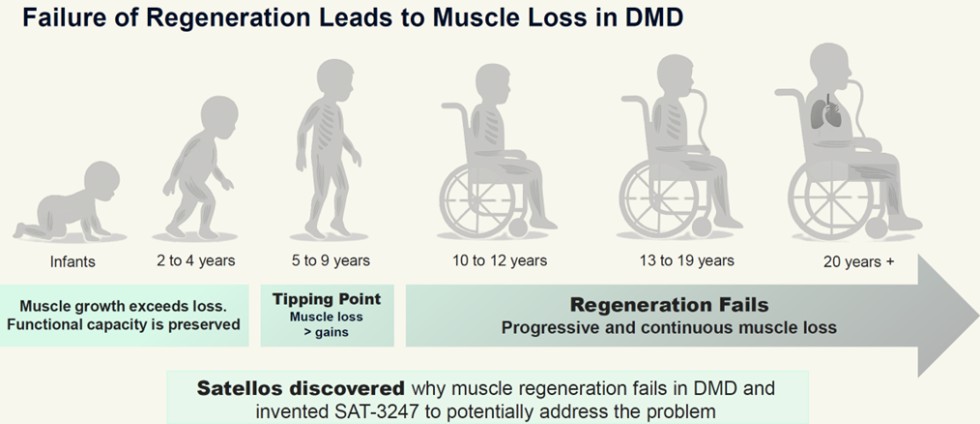
Figure 2: Progressive muscle loss a hallmark of Duchenne muscular dystrophy.
Duchenne is caused by a mutation in the dystrophin gene that results in impairment to or loss of the dystrophin protein. Dr. Rudnicki demonstrated that muscle stem cells require a signal from the dystrophin protein to properly and efficiently divide in an asymmetric fashion (Source: Dumont et al. 2015, Nature Medicine.). As described in Figure 3 below, without the dystrophin signal, muscle stem cells fail to divide efficiently, often creating copies of themselves rather than making the progenitor cells needed to create new muscle.
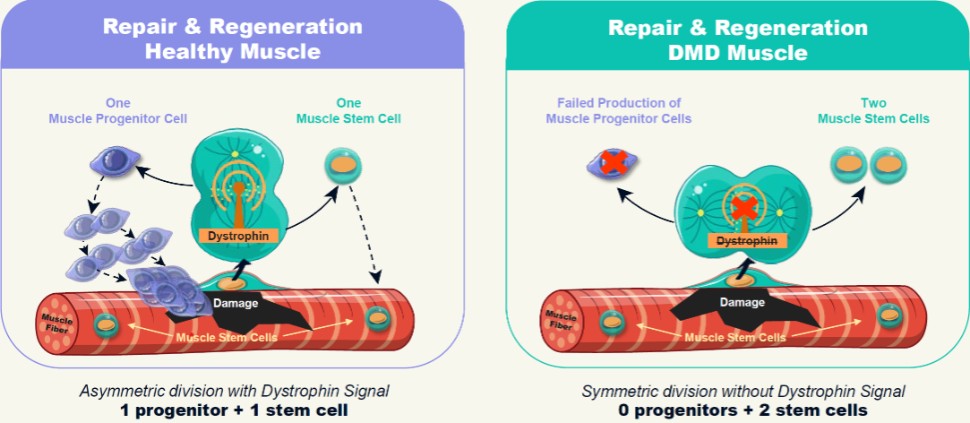
Figure 3: Imbalanced Stem Cell Division
To address this problem, Satellos’ therapeutic strategy is to restore the missing signaling role of dystrophin by drug treatment - thereby resetting the muscle regeneration process.
- 15 -
Restoring the Missing Dystrophin Signal via AAK1 Inhibition
Deploying MyoReGenX™ to build on the identification and discovery of this previously unreported signaling role of dystrophin, in collaboration with the Rudnicki lab, Satellos undertook a systematic assessment, evaluation and prioritization of molecular pathways for their potential to safely rescue asymmetric stem cell divisions in the absence of dystrophin. From this exercise conducted over a multi-year period, the Company identified and selected Adaptor Associated Kinase 1 (aka “AAK1”), a protein kinase in the molecular signaling pathway known as “Notch”. Satellos has generated extensive preclinical data in the Mdx mouse, a gold standard research model bearing the same genetic defect as patients with Duchenne, demonstrating that treatment of these research mice through inhibition of AAK1 with SAT-3247 has potential to restore the process of asymmetric division in muscle stems cells. Our preclinical studies have further shown that inhibition of AAK1 with SAT-3247 enables muscle regeneration with the potential to increase muscle strength. Thus, we believe, SAT-3247 represents a potential novel therapeutic drug for the treatment of Duchenne in humans. Figure 4 below depicts our understanding of the mechanism by which our lead drug candidate, SAT-3247, affects asymmetric division and the muscle stem cell mediated regeneration via inhibition of AAK1.
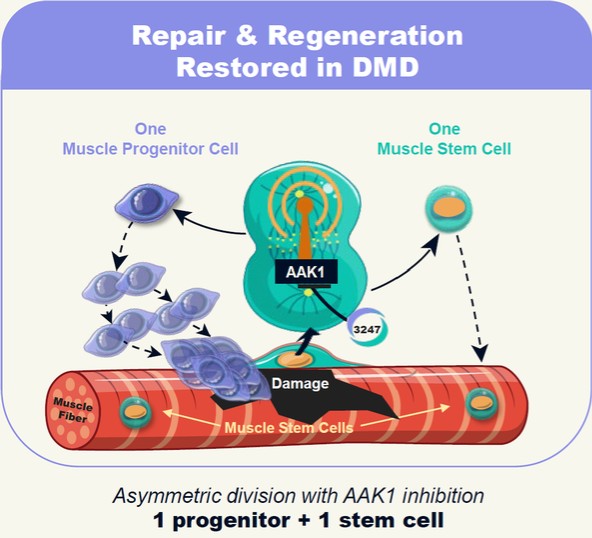
Figure 4: Satellos Approach: Reset regeneration with SAT-3247
Small molecule inhibitors of AAK1 have previously been described for non-muscle related disease indications by Lexicon Pharamceuticals Inc., an unrelated biotech company, which has reported what appears to be acceptable safety profiles in multiple human clinical trials spanning hundreds of patients. We believe this provides some initial indications of the potential safety of AAK1 inhibition.
Satellos announced positive preclinical data presented at the March 2024 MDA Clinical and Scientific Conference showing that SAT-3247 can improve skeletal muscle function in multiple mouse models of muscle degeneration. The preclinical data presented show the broad potential of SAT-3247 to improve skeletal muscle function as it has been demonstrated in three mouse models of muscle degeneration: mdx model of Duchenne, FLExDUX4 model of FSHD, and a muscle injury model in wildtype mice. In all instances, treatment with SAT-3247 over a three-to-four-week period resulted in a statistically significant improvement in muscle force versus animals receiving placebo.
In October 2024, Satellos announced data presented at the 29th Annual Congress of the World Muscle Society in Prague. The data presented from the open-label pilot study demonstrated that treatment of two DMD canines with SAT-3247 improved measures of strength to near normal levels.
- 16 -
The Company has filed for patent protection on SAT-3247 and other inhibitors of AAK1. Please refer to the Intellectual Property section in the Company’s AIF for further details on its intellectual property strategy and filings and its licensing agreement with the OHRI.
Drug Discovery and Development: Infrastructure
Satellos is building on its science and discoveries to create the first ever therapy intended to regulate muscle stem cell driven regeneration capacity and, potentially, address what we believe to be a potential root cause of the progressively degenerating nature of Duchenne. Specifically, our goal is the development of a first-in-class, small molecule orally administered drug.
To achieve this goal, Satellos has established an in-house team of drug development professionals to oversee its workplan and manage its infrastructure. This includes personnel with expertise in pre-clinical development, chemistry, manufacturing and controls (“CMC”) and clinical development. This in-house team is focused on managing a network of specialized contract research organizations (“CROs”). We rely on these CRO’s to complete, under our supervision, the pre-clinical and IND enabling studies, CMC activities as well as execution of the planned clinical development program.
In addition to our internal team, we have outsourced ongoing research discovery efforts under our agreement with OHRI.
Current Status - Summary
The Company is currently conducting two clinical trials with SAT-3247 known as TRAILHEAD and BASECAMP which have been previously described above.
Follow-On Program
There are more than 30 types of muscular dystrophy that affect humans. Each of these dystrophies has different causes that manifest into conditions ranging in severity from benign, small impairments to motor function, to the full loss of ambulation, or even death. Satellos has conducted proof of concept preclinical studies in relevant animal disease models showing potential for benefit by restoring the muscle regeneration process in Lama-2 Related Muscular Dystrophy (prevalence estimates between one in 50,000 and one in 400,000 births), Collagen-VI Related Muscular Dystrophy (prevalence of severe form of the disease estimated to be one in 1,000,000 births) and Facioscapulohumeral muscular dystrophy (“FSHD”) (prevalence of 4 per 100,000 individuals). These represent potential follow-on disease indications or programs for Satellos to consider in the future. The Company also plans to evaluate additional dystrophies as part of its ongoing research and development efforts.
The Company has developed a plan to initiate a Phase 2 clinical trial in FSHD. The intended objectives and endpoints of the study would include safety, drug concentration, fluid-based biomarkers and efficacy. The Company anticipates that the study would enroll approximately 50 adult participants with FSHD. The precise protocol of the study is under development. The Company currently anticipates filing the necessary regulatory documents to initiate a Phase 2 clinical trial in the United States and Canada in the second quarter of 2026.
Preferential Programs
As a developer of therapeutics to treat a number of rare diseases, Satellos intends to apply for specific US government sponsored development programs that have potential in certain circumstances to accelerate approval timelines and enhance market exclusivity. These programs include (but may not be limited to):
| (1) | Orphan Drug Designation |
Under the Orphan Drug Act, the FDA may grant Orphan Drug Designation to a drug intended to treat a rare disease or condition, which is defined as one affecting fewer than 200,000 individuals in the United States or more than 200,000 individuals where there is no reasonable expectation that the product development cost will be recovered from product sales in the United States. Orphan Drug Designation must be requested before submitting a new drug application (“NDA”). More than one product candidate may receive an Orphan Drug Designation for the same indication. Orphan Drug Designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
- 17 -
Benefits for drugs that are bestowed ‘orphan status’ may include tax credits on clinical testing, waiving of the NDA application fee, and potential eligibility for a seven-year market exclusivity upon approval of the drug. In particular, if an orphan drug-designated drug subsequently receives FDA approval for the disease for which it was designed, the drug will be entitled to seven years of product exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication, except in very limited circumstances (such as a showing of clinical superiority to the product with orphan exclusivity by means of greater effectiveness, greater safety, or providing a major contribution to patient care or in instances of drug supply issues), for seven years. Orphan exclusivity does not block the approval of a different drug for the same rare disease or condition, nor does it block the approval of the same drug for different conditions. If a competitor obtains approval of the same drug, as defined by the FDA, or if our product candidate is determined to be the same drug as a competitor’s product for the same indication or disease, the competitor’s exclusivity could block the approval of our product candidate in the designated orphan indication for seven years, unless our product is demonstrated to be clinically superior to the competitor’s drug.
A product with Orphan Drug Designation may not receive orphan exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
On August 8, 2024, Satellos announced that the FDA granted Orphan Drug Designation to SAT-3247 for the treatment of Duchenne muscular dystrophy.
| (2) | Rare Pediatric Disease Designation |
The FDA also has authority to award priority review vouchers to sponsors of certain rare pediatric disease product applications. This program is designed to encourage development of new drugs for the prevention and treatment of certain rare pediatric diseases. Specifically, under this program, a sponsor who receives an approval for a drug for a “rare pediatric disease” may qualify for a voucher that can be redeemed to receive a priority review of a subsequent marketing application for a different product. The sponsor of a rare pediatric disease drug product receiving a priority review voucher may transfer (including by sale) the voucher to another sponsor. The voucher may be further transferred any number of times before the voucher is used, as long as the sponsor making the transfer has not yet submitted the application. The FDA may also revoke any priority review voucher if the rare pediatric disease drug for which the voucher was awarded is not marketed in the United States within one year following the date of approval.
A sponsor may request that FDA designate a product candidate as being intended for a “rare pediatric disease” before it submits an NDA for the product, including at the same time that Orphan Drug Designation is requested. Designation of a product candidate as a product for a rare pediatric disease does not guarantee that a marketing application for such product candidate will meet the eligibility criteria for a priority review voucher at the time the application is approved
For purposes of this program, a “rare pediatric disease” is a (a) serious or life-threatening disease in which the serious or life-threatening manifestations primarily affect individuals aged from birth to 18 years; and (b) rare diseases or conditions within the meaning of the Orphan Drug Act. Congress has only authorized the Rare Pediatric Disease Priority Review Voucher Program until September 30, 2029. Consequently, unless Congress reauthorizes the program, the sponsor of the marketing application for a drug that receives Rare Pediatric Disease Designation will only be eligible to receive a voucher if the FDA grants the voucher on or before September 30, 2029.
On August 8, 2024, Satellos announced that the FDA granted a Rare Pediatric Disease Designation to SAT-3247 for the treatment of DMD.
| (3) | Accelerated Approval |
A drug product may also be eligible for accelerated approval if it is designed to treat a serious or life-threatening disease or condition and provides a meaningful therapeutic benefit over existing treatments. Such product candidates can be approved upon a determination that the product candidate has an effect on either a surrogate endpoint that is reasonably likely to predict clinical benefit or on an intermediate clinical endpoint that can be measured earlier than irreversible morbidity or mortality, or IMM, that is reasonably likely to predict an effect on IMM or other clinical benefit, taking into account the severity, rarity, or prevalence of the disease or condition and the availability or lack of alternative treatments.
- 18 -
As a condition of approval, the FDA generally requires that a sponsor of a drug receiving accelerated approval perform adequate and well-controlled confirmatory clinical trials to verify and describe the predicted effect on IMM or other clinical endpoint, and may require that such confirmatory trials be underway prior to granting accelerated approval. The FDA may withdraw approval of a drug or indication approved under accelerated approval on an expedited basis if, for example, the confirmatory trial fails to verify the predicted clinical benefit of the product or the sponsor fails to conduct such confirmatory trials in a timely manner or in accordance with applicable regulations. Drugs granted accelerated approval must meet the same statutory standards for safety and effectiveness as those granted traditional approval. In addition, all promotional materials for products approved under the accelerated approval program are subject to prior review by the FDA.
It is unknown at this time whether the Company’s product candidate will be eligible for accelerated approval.
Regulatory Process
Government authorities in the United States, including federal, state, and local authorities, and in other countries, extensively regulate, among other things, the manufacturing, research and clinical development, marketing, labeling and packaging, storage, distribution, post-approval monitoring and reporting, advertising and promotion, and export and import of biological products, such as those we are developing. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. Securing final regulatory approval for the manufacture and sale of drug products in the US, Europe, Canada and other commercial territories, is a long and costly process that is controlled by that particular territory’s regulatory agency. The regulatory agency in the US is the FDA, in Canada it is Health Canada, and in Europe it is the European Medicines Agency. Other regulatory agencies have similar regulatory approval processes, but each regulatory agency has its own approval processes. Approval in the US, Canada or Europe does not assure approval by other regulatory agencies, although often test results from one country may be used in applications for regulatory approval in another country.
Satellos has completed a Phase 1a/b clinical trial with SAT-3247 in healthy volunteers and adults with DMD. The company has also initiated TRAILHEAD, an open-label study in adult participants, and BASECAMP, a Phase 2 pediatric study. The time required to obtain marketing approval by regulatory authorities is unpredictable but typically takes many years following the commencement of preclinical studies and clinical trials and will require significant additional capital.
US Government Regulations
In the US, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (“FDCA”) and its implementing regulations. FDA approval is required before any new unapproved drug or dosage form, including a new use of a previously approved drug, can be marketed in the United States. Drugs are also subject to other federal, state, and local statutes and regulations. If we fail to comply with applicable FDA or other requirements at any time during the product development process, clinical testing, the approval process or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, and civil monetary penalties or criminal prosecution. Any FDA enforcement action could have a material adverse effect on us. The process required by the FDA before product candidates may be approved for marketing in the United States generally involves the following:
| ● | completion of nonclinical laboratory tests, animal studies, and formulation studies in accordance with FDA’s good laboratory practice (GLP) requirements and other applicable regulations; |
| ● | submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| ● | approval by an independent institutional review board (IRB)/ethics committee (EC), either centralized or with respect to each clinical site, before each clinical trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials in accordance with good clinical practice (GCP) requirements to establish the safety and efficacy of the proposed drug for its intended use; |
| ● | submission to the FDA of a New Drug Application (NDA) after completion of necessary and applicable supportive and registrational clinical trials; |
- 19 -
| ● | determination by the FDA within 60 days of its receipt of an NDA to accept the filing for substantive review; |
| ● | satisfactory completion of an FDA advisory committee review, if determined necessary by the agency; |
| ● | FDA inspection, if requested, of the manufacturing facility or facilities at which the drug is produced to assess compliance with cGMP requirements to ensure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality, and purity, and of selected company locations or clinical investigation sites to assess compliance with GCP guidelines, protocol adherence, and data integrity; and |
| ● | FDA review and approval of the NDA to permit commercial marketing of the product for particular indications for use in the United States. |
Accordingly, Satellos has submitted an IND to the FDA for its clinical trials for SAT-3247 which is currently effective. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of in vitro or in vivo preclinical studies assessing the toxicology, PK, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data such as from prior clinical trials in another regulatory jurisdiction, or any relevant literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold until the IND sponsor and the FDA resolve any outstanding concerns or questions. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with the International Conference on Harmonization GCP guidelines, which include the requirement that all research subjects, or parent/legal guardians of research subjects if appropriate for the population studied, provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, a central IRB or an independent IRB for each site proposing to conduct the clinical trial must review and approve the protocol for any clinical trial and its informed consent form before the clinical trial begins at that site and must provide ethical oversight for the study until it’s completed. Some trials also include oversight by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board, which may review data and endpoints at designated check points, make recommendations, and/or halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical trial results to public registries (e.g., clinicaltrials.gov).
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
Phase 1: The product candidate is initially introduced into healthy human subjects or patients with the target disease or condition. These studies are designed to test the safety, dosage tolerance, absorption, metabolism, and distribution of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy human subjects, the initial human testing is often conducted in patients.
Phase 2: The product candidate is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages, and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials.
Phase 3: The product candidate is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval.
- 20 -
Post-approval clinical trials, sometimes referred to as Phase 4 studies, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA.
The FDA or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. In addition, some clinical trials are overseen by an independent group of qualified experts organized by the sponsor, known as a data safety monitoring board or committee. Depending on its charter, this group may determine whether a clinical trial may move forward at designated check points or when particular criteria are met, based on access to certain data from the clinical trial.
During the development of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase 2, and before an NDA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and the FDA to reach agreement on the next phase of development. Sponsors typically use the meetings at the end of the Phase 2 clinical trial to discuss Phase 2 clinical results and present plans for the pivotal Phase 3 clinical trials that they believe will support approval of the new drug. Fast Track Status and Breakthrough Designation provide opportunities for sponsor to have more frequent interaction with the FDA.
Phase I, Phase II, and Phase III clinical testing may not be completed successfully within a specified period, if at all, and there can be no assurance that the data collected will support FDA approval of a product candidate. Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality, and purity of the final drug. In addition, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
While the IND is active, progress reports summarizing the results of the clinical trials and nonclinical studies performed since the last progress report must be submitted at least annually to the FDA. Written IND safety reports must also be submitted to the FDA and investigators for serious and unexpected suspected adverse events, findings from other studies suggesting a significant risk to humans exposed to the same or similar drugs, findings from animal or in vitro testing suggesting a significant risk to humans, and any clinically important increased incidence of a serious suspected adverse reaction compared to that listed in the protocol or investigator brochure.
NDA Review and Approval Process
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development nonclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product. The submission of an NDA is subject to the payment of substantial user fees; a waiver of such fees may be obtained under certain limited circumstances. Additionally, no user fees are assessed on NDAs for products designated as orphan drugs, unless the product also includes a non-orphan indication. Congress is required to re-authorize the agency’s user fee programs every five years, and current legislative provisions supporting the prescription drug-specific program are set to expire on September 30, 2027.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing; in this event, the NDA must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Under the Prescription Drug User Fee Act (PDUFA) guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes 12 months from the date the NDA is submitted to FDA because the FDA has approximately two months to make a “filing” decision after the application is submitted.
- 21 -
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect a sponsor location and one or more clinical sites to assure compliance with GCP guidelines. If the FDA determines that the application, manufacturing process, manufacturing facilities, or conduct of the clinical trials are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The approval process is lengthy and often difficult, and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data and information. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant to assure and preserve the product’s identity, strength, quality, and purity. Based on the FDA’s evaluation of an NDA and all supporting information, including the results of manufacturing facility inspections, it will issue either an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete, and the application will not be approved in its present form. A Complete Response Letter usually describes the specific deficiencies in the NDA identified by the FDA and may require additional clinical data, such as an additional pivotal Phase 3 clinical trial or other significant and time-consuming requirements related to clinical trials, nonclinical studies, or manufacturing. In September 2025, the FDA began publishing Complete Response Letters soon after issuing them to the respective sponsors, breaking with long standing agency tradition of publishing Complete Response Letters with approval documentation only after the product candidate is approved. If a Complete Response Letter is issued, the sponsor may choose either to resubmit the NDA, addressing all of the deficiencies identified in the letter, or to withdraw the application. Even if such data and information are submitted, the FDA may decide that the NDA does not satisfy the criteria for approval. If and when all deficiencies have been addressed to the FDA’s satisfaction in a resubmitted NDA, the FDA will issue an approval letter.
If regulatory approval of a product is granted, such approval will be granted for particular indications and may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the NDA with a risk evaluation and mitigation strategy (REMS) to ensure the benefits of the product outweigh its risks. A REMS is a safety strategy to manage a known or potential serious risk associated with a medicine and to enable patients to have continued access to such medicines by managing their safe use. It could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries, and other risk minimization tools. The FDA also may offer accelerated approval with postmarketing confirmatory trial requirements or approvals subject to other postmarketing requirements. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing requirements is not maintained or if problems occur after the product reaches the marketplace. The FDA may also require one or more Phase 4 post-marketing studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization and may limit further marketing of the product based on the results of these post-marketing studies. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could impact the timeline for regulatory approval or otherwise impact ongoing development programs.
Intellectual Property: Satellos Portfolio
Intellectual property, including patents, trade secrets, trademarks and copyrights, is important to our business. Our commercial success depends in part on our ability to obtain and maintain proprietary intellectual property protection for our current product candidate, SAT-3247, as well as for future product candidates and novel discoveries, product development technologies, and know-how. Our commercial success also depends in part on our ability to operate without infringing on the proprietary rights of others and to prevent others from infringing our proprietary rights. We seek to maintain our proprietary position by, among other means, filing United States and foreign patent application to obtain issued patents that cover our product candidates, technology, inventions, and improvements that are important to the development and implementation of our business. We also license from third parties certain patent rights and proprietary know-how that we believe to be necessary or useful to our business. Additionally, we protect our proprietary know-how that may not be patentable, and other confidential information, by maintaining and implementing appropriate policies and procedures for ensuring secrecy and confidentiality.
- 22 -
Our patent portfolio is built with a goal of establishing broad protection that generally includes claims directed to the composition of matter of our product candidates, pharmaceutical compositions or formulations, salts and solid forms of our product candidates, methods of synthesis, and methods of treatment using our product candidates. We are currently seeking and maintaining patent protection in the United States and key foreign jurisdictions, or intend to seek and maintain patent protection in key foreign jurisdictions, where we intend to market our product candidates, and plan to do so with respect to any of our future product candidates. Our patent portfolio includes a combination of patents and pending patent applications solely owned by us and patents and pending patent applications licensed from the OHRI.
As of March 27, 2026, our patent estate contains 12 patent families comprising 3 issued U.S. patents, 18 issued patents in Canada, France, Germany, Italy, Japan, Spain, Switzerland, and the United Kingdom, 8 pending U.S. non-provisional patent applications, and 22 pending patent applications in various jurisdictions outside of the U.S., which we own or exclusively license.
With regard to SAT-3247, we own 3 pending U.S. non-provisional patent applications, and pending patent applications in Australia, Brazil, Canada, China, Europe, India, Israel, Japan, South Korea, Mexico, New Zealand, Saudi Arabia, Singapore, Taiwan, United Arab Emirates, and South Africa relating to SAT-3247 composition of matter, which if issued are expected to expire in 2044. We also own 1 pending U.S. non-provisional patent application, 1 pending PCT international application, and 1 pending patent application in Taiwan relating to salts and solid forms of SAT-3247, which, if issued, are expected to expire in 2045. We further own 2 patent families directed to methods of treating muscular dystrophy with SAT-3247, with 1 pending U.S. non-provisional patent application, 1 pending PCT international application, and 5 pending U.S. provisional applications, which, if issued, are expected to expire between 2045 and 2046. The foregoing expiration dates do not account for potentially available patent term adjustments or extensions and terminal disclaimers, and assume payment of all appropriate maintenance, renewal, and annuity fees.
Further, we have and will continue to pursue trademark protection for our company SATELLOS and other brands. As of February 2, 2026, we own 55 registered trademarks in foreign jurisdictions and 6 pending trademark applications in the United States and foreign jurisdictions.
License Agreement: Ottawa Hospital Research Institute
Effective May 1, 2018, Satellos and the OHRI entered into a license agreement whereby the OHRI granted Satellos an exclusive, world-wide, sublicensable, royalty bearing right and license to a body of technology and patents comprised of five patent families to develop, make, have made, import, use, offer for sale, sell and have sold or otherwise commercialize licensed products (the “OHRI License”). The OHRI License contains provisions describing a process for the ongoing disclosure and potential licensing to Satellos of new discoveries or inventions which may occur at the OHRI and which may have relevance to the field in which the Company operates as it is defined in the OHRI License.
Ottawa Hospital Research Institute
Effective May 1, 2018, Satellos and the OHRI entered into a sponsored research agreement, during the term of which OHRI has agreed to carry out specific research and development activities according to a prescribed statement of work, as may be amended from time to time, under the direction of the Company’s co-founder, Dr. Michael A. Rudnicki (the “OHRI SRA”).
Under the OHRI SRA, inventions developed solely by OHRI researchers in the course of the research program are owned by OHRI. Satellos holds an exclusive option to obtain an exclusive license to such inventions under the parties’ license agreement. Inventions jointly developed by OHRI researchers and Satellos personnel are co-owned by the parties.
Competitive Conditions
The biotechnology industry which the Company operates within is intensely competitive in all its phases, and the Company faces immense competition from other companies, some of which can be expected to have more financial resources and retail, formulation, research, processing, and marketing experience than the Company. There can be no assurance that potential competitors of the Company, which may have greater financial, formulation, research, production, sales and marketing experience, and personnel and resources than the Company, are not currently developing, or will not in the future develop, products and strategies that are equally or more effective and/or economical as any products or strategies developed by the Company or which would otherwise render the Company’s business, products and strategies, as applicable, ineffective, or obsolete. Increased competition by larger and better- financed competitors could materially and adversely affect the business, financial condition and results of operations of the Company.
- 23 -
Market Overview
Since its inception, Satellos’ lead therapeutic program has been acutely focused on the discovery and development of novel, disease modifying treatments for the lethal pediatric muscle disease, DMD. DMD is a progressive genetic muscle wasting medical condition that is predominantly found in boys. Although spontaneous gene mutations can occur, Duchenne is primarily an inherited X-linked genetic disorder whereby a mutation in the dystrophin gene results in the absence or the severely impaired production of the dystrophin protein which is essential for muscle structural integrity and the ability of muscle stem cells to properly regenerate injured muscles. To date, Duchenne remains uncurable.
Duchenne is the most severe form of dystrophinopathies and presents as a progressive weakening and degeneration of the heart, respiratory muscles and skeletal muscles in children. The first symptoms of muscle weakness are typically noticed between the ages of 3 – 5 years and worsen with age to the point of a loss of ambulation. With continued degeneration and loss of function, the condition causes pre-mature death via cardiovascular complications. The National Center for Advancing Translational Sciences, part of the National Institutes of Health, estimates that the combined Duchenne patient population across all ages approaches 300,000 persons globally. The current standard of care and the main focus of investigative research has been directed toward slowing the progression of the disease or managing its symptoms.
Current treatments for Duchenne are classified under three main areas: glucocorticoids (steroids), oligonucleotide medicines and gene therapies.
The use of corticosteroid or corticosteroid-derived medicines prednisone (1974), branded EMFLAZA® (deflazacort, 2017) and more recently AGAMREE® (vamorolone, 2023), represent the standard of care in the treatment of Duchenne. These medicines have largely been responsible for quality-of-life improvements since their introduction and have played an important role in alleviating the inflammation associated with this disease leading to improvements in muscle strength, a slowing of muscle loss and an extension of ambulation. PTC Therapeutics’ revenue for deflazacort was US$146.4 million in 2025 (Source: PTC Therapeutics) with an annual cost of approximately US$80,000 per patient (Source PMID: 32223597) ; Deflazacort has also been associated with a significant side effect profile and a failure to treat the fundamental cause of the dystrophinopathy. Santhera Pharmaceuticals launched AGAMREE® in Germany in January 2024, while its North American partner, Catalyst Pharmaceuticals initiated its launch of vamorolone in mid-March 2024. Based on available retail price estimates, yearly costs could reach as high as US$291,000 for patients weighing 50kg and above who take the recommended dose of 6mg/kg daily (Source: Drugs.com). Revenue for AGAMREE® was US$117.1M for 2025.
A recently approved treatment approach focused on very specific genetic mutations found within the dystrophin gene is called exon-skipping. In this regard, exon-skipping therapies seek to skip or pass over a mutated region of the dystrophin gene as it is being read which facilitates some production of the dystrophin protein. This class of drug is specific to the patient’s gene mutation, for instance, Exon 45 mutations specifically require Exon 45 skipping and Exon 45 skipping therapies will not work in a patient with a mutation of Exon 51 or Exon 53 for example.
While evidence of clinical improvement is minimal, Sarepta Therapeutics’ approved exon-skipping drug therapies, EXONDYS 51 (eteplirsen, 2016), VYONDYS 53 (golodiresen, 2019) and AMONDYS 45 (casimersen, 2021) generated approximately US$965.6M in 2025. Annual costs for exon-skipping therapy are approximately US$750,000 (Source: Drugs.com). These therapies are only indicated for patients with exon 51, 53 and 45 mutations, which represent approximately 12%, 10% and 9% of the Duchenne population, respectively. A competing product, Exon 51-skipping Viltepso® (viltolarsen) from NS Pharmaceuticals, was approved in the U.S. in 2020 and generated US$130 million in revenue in 2025 (Source: Nippon Shinyaku) based on estimated annual per patient cost of US$733,000 for a 30kg patient (Source: viltepso-new-drug-fact-blast.pdf).
Elevidys (delandistrogene moxeparvovec) was approved by the FDA in June 2023 within the gene therapy segment. for the treatment of ambulatory Duchenne patients aged 4 through 5 years old, Elevidys’. Developed and marketed by Sarepta Therapeutics, Elevidys is a gene therapy product that encodes and increases the production of micro-dystrophin in individuals with Duchenne. FDA approval was based on the expectation that an increased expression of micro-dystrophin was “reasonably likely to predict clinical benefit” in DMD patients. In 2024, the label was broadened to all patients with a confirmed diagnosis of DMD, including non-ambulatory individuals. Due to safety concerns in 2025, the label was then restructured to only reflect ambulatory patients over the age of 4. Sales of Elevidys generated US$898.7 million in revenues in 2025 (Source: Sarepta.com). The price of the one time dose administration of Elevidys is US$3.2 million.
- 24 -
On March 21, 2024, the FDA granted marketing approval to Italfarmaco SpA’s givinostat (Duvyzat) for individuals with DMD aged 6 years and older. Duvyzat is the first non-steroidal drug approved to treat patients with all genetic variants of DMD. It has been designed to inhibit histone deacetylase enzymes. In the phase 3 EPIDYS trial which provided the supporting data for approval, results showed a slower decline in treated patients on the time to climb 4 stairs in comparison with a placebo group. We believe the approval of a non-steroidal drug potentially suitable for Duchenne patients regardless of dystrophin mutation status is a positive development for patients and a favourable portent for Satellos’ development and commercialization planning.
Despite efficacy considerations, growth estimates by market analysts remain quite high, with an estimated compound annual growth rate of ~17% (Source: Fortune Business Insights) for these and other products currently in late-stage development for DMD. Based on approved product sales data in the major global pharmaceutical markets, the current market for therapeutics to treat Duchenne is estimated to be US$4.6B in 2025. The projected growth estimates for both current and expected new therapeutics intended to be disease modifying, such as micro-dystrophin gene replacement therapies, is projected to reach US$19 B by 2035 (Source: Fortune Business Insights). It is believed that these favourable forecasts are due to the lack of effective treatment options and the understandable longing of patients and their families for relief from this debilitating, progressive and ultimately fatal disease.
Notwithstanding the significant market projections, all current therapeutic approaches, either approved or currently in development, including exon skipping, gene correction and gene therapy, predominantly aim to enhance the function and preservation of existing muscle tissues. As a result, these approaches are unlikely to be effective at addressing the inherent regeneration deficits seen in DMD patients and identified by Satellos which we believe is a root cause explaining the progressive, ongoing muscle loss that is experienced by people living with Duchenne. Satellos’ approach, in attempting to reinstate the ability of muscle to regenerate through the specific pharmaceutical targeting of muscle stem cells, is unique. It also, being dystrophin-independent and not tied to specific mutations, has potential to be a stand-alone therapy across a broad spectrum of ambulatory and non-ambulatory patients as well as add-on therapy to other approaches which do not address regeneration.
Based on the potentially transformative and disease modifying nature of treating a root cause of Duchenne, Satellos anticipates significant market potential for its products. It is estimated that a novel chronically delivered therapeutic with disease modifying benefits, such as the drug Satellos envisages, could be priced in excess of US$750,000 per patient annually. This conservative estimate is based on referencing the treatment cost of the exon skipping drugs previously mentioned, yet with the ability to improve muscle function rather than simply halt its decline. Based on these estimates, and a significant global patient pool, the annual revenue potential for an effective, disease modifying drug treatment for Duchenne could exceed $1 billion per year.
Given the precedent of cost approvals of competing DMD drugs, Satellos believes that the patient per year costs will be covered by insurance providers and/or national health systems. Our expectation remains that, based on its molecular mechanism of action, our future drug product has the potential to confer several advantages over existing treatment approaches, including reduced untoward side effects (e.g., diabetes, bone damage, immune reactions), ease of administration (tablet), and functional benefits from the repair and regeneration of muscle. Thus, with an improved quality of life and an increased efficiency of care resources, we believe the arguments in favour of general reimbursement for this drug have the potential to be quite strong.
Business Strategy
To advance SAT-3247 and potentially accelerate regulatory approvals, Satellos aims to first demonstrate safety, biological activity and the potential for functional benefit through the treatment of Duchenne patients in different age groups in Phase 1 (completed) and Phase 2 clinical trials (underway as TRAILHEAD and BASECAMP). In parallel, we intend to leverage our existing orphan drug and rare pediatric disease designations as well as, data supporting, apply for ‘accelerated approval’ and ‘breakthrough status’ to expedite approval of SAT-3247. We believe this represents, collectively, a sound basis on which to create shareholder value and market demand for our products and technologies. It will inform and enable our ability to explore and evaluate three broad approaches for optimizing value creation and monetization: (1) leveraging market demand into development and commercialization partnerships with biopharmaceutical companies in risk-reward sharing arrangements; (2) continuing to advance development and commercialization of our lead clinical product for Duchenne and other indications; and (3) exploring M&A transactions. The foregoing may include a mix of joint venture arrangements, regional partnerships or co-development and co-marketing collaborations, depending on clinical trial outcomes, market and environmental conditions, and risk/reward trade-offs. Through these relationships, Satellos will seek to have its products approved and registered for sale globally.
- 25 -
Specialized Company Skill and Knowledge
The Company intends to establish itself as a leader in the research and development of novel therapeutics for the treatment of life-threatening diseases. The Company’s directors and officers possess a breadth of knowledge and expertise to develop and execute the Company’s business strategy and support its technical capabilities. In addition, by leveraging the management and operational team’s combined expertise, the Company believes it possesses the ability to execute its commercial and operational strategy. The Company intends to continue to build-out its team with additional specialists and expert consultants to align with the core business goals and objectives in a time-sensitive and cost-effective manner. The Company’s full and part-time employees and key consultants include 8 individuals with PhD’s, 1 with an M.D., 6 with an MBA or MSC, and 5 with a finance designation.
Business Cycles
The Company’s business is not expected to be cyclical or seasonal.
Economic Dependence
The Company’s business is not expected to be economically dependent on any contracts for the foreseeable future.
Changes to Contracts
Satellos does not reasonably anticipate being materially affected by re-negotiation or termination of contracts or sub-contracts.
Environmental Protection
There are no financial and operational effects of environmental requirements on the capital expenditures, profit or loss and competitive position of the Company in the current financial year and none is expected in future years.
Employees
As of December 31, 2025, Satellos, directly and through its subsidiaries, employed eighteen full-time employees, three part-time employees and ten key consultants in Canada and the United States.
Dr. Michael Rudnicki is not an employee, but, pursuant to a consulting contract, provides key services to the Company in the role of Chief Discovery Officer. The Company also contracts with other consultants on an as-needed basis to provide highly skilled and specialized knowledge in a cost-efficient manner.
Foreign Operations
The Company has two wholly-owned subsidiaries, both of which are foreign: an Australian subsidiary, Satellos Bioscience Australia Pty Ltd. and a United States (Delaware) subsidiary, Satellos Bioscience US, Inc.
Social or Environmental Policies
To date, the Company has not implemented social or environmental policies. However, the Company is aware that it may need to adopt additional social, environmental and human rights policies in the future.
Reorganizations
The Company has not completed any material reorganization within the three most recently completed financial years and no material reorganization is currently proposed.
- 26 -
Legacy Asset
Through its business combination with iCo the Company acquired certain assets. Amphotericin B Technologies Inc. (“Amp B”) was established as a wholly owned subsidiary of the Company as part of a renewed strategy to create value and attract partnerships and/or funding.
On October 6, 2022, NW Micelle Therapeutics Inc. (“NWMT”) was established with NW PharmaTech holding 85% of the shares in NWMT and Amp B holding 15%.
On July 29, 2025, the Company completed the sale of AmpB, to Natural Works Trading Limited, as part of a strategic portfolio review, for consideration of US$10 thousand. This divestiture did not impact the Company’s core operations or ongoing business activities and the Company has no further involvement or obligation related to these legacy assets.
- 27 -
Investing in our securities involves a high degree of risk. Before deciding to invest in our securities, you should carefully consider the risks described below, together with other information included in or incorporated by reference into this Annual Information Form and filed on SEDAR+ at www.sedarplus.ca. If any of the following risks materialize, the business, financial condition, results of operations and future prospects of the Company will likely be materially and adversely affected. This could cause actual future events to differ materially from those described in forward-looking statements and may cause the trading price of our securities to decline. The following discussion highlights some of the risks and uncertainties facing the Company.
Risk Factors Relating to the Company
The Company’s principal business is substantively constituted on the discovery and development of new drug candidates to treat muscle wasting disorders, initially for Duchenne. Due to the nature of Satellos’ business as a biotechnology company, the Company may be subject to significant risks and uncertainties, including but not necessarily limited to risks and uncertainties related to technical and scientific, regulatory and clinical, and business and financial matters, which are set out and discussed below. Readers should carefully consider all such risks and uncertainties set out in the discussion below and be mindful that additional risks and uncertainties not currently known or reasonably foreseeable may arise.
Satellos may not be able to maintain our operations and research and development without additional funding, and we may not have access to sufficient capital.
Since its inception, Satellos has incurred significant losses and we have financed our operations primarily through public offerings of Common Shares, private placements, the issuance of convertible notes and research tax credits. We have incurred significant operating losses and negative cash flows from operations since inception. As of December 31, 2025 we had available cash, cash equivalents and short-term investments totaling US$27.7 million. Subsequent to the year end, on February 9, 2026, we completed an underwritten public offering for gross proceeds of approximately US$57.2 million. Based on management’s estimate and current level of operations, we believe that our current liquidity position is sufficient to finance our operations through 2027. We will need to raise additional capital to fund our operations and to develop SAT-3247 and our other development candidates. Our future capital requirements will be substantial and may increase beyond current expectations depending on many factors, such as the duration, scope, rate of progress, results and costs of any preclinical studies and clinical trials for our current or any future product candidates; unexpected delays or developments in seeking regulatory approvals and the outcome thereof; the time and cost in preparing, filing, prosecuting, maintaining, and enforcing patent claims; other unexpected developments encountered in implementing our business development and commercialization strategies; the outcome of any litigation; and arrangements with collaborators. Further, changing circumstances may cause us to consume capital significantly faster than we currently anticipate. We have based the foregoing estimates on assumptions that may prove to be wrong, and we could utilize our available financial resources sooner than we currently expect.
We may seek to raise additional funds through public or private equity or debt financing, collaboration agreements with other companies and/or from other sources. We have no committed source of additional capital and additional funding may not be available on terms that are acceptable to us, or at all. If adequate funding is not available on reasonable terms, we may need to obtain funds on terms less favourable than we would otherwise accept. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the issuance of those securities could result in dilution to our shareholders. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of SAT-3247 or other future product candidates or other research and development initiatives. We could be required to seek collaborators for our product candidates at an earlier stage than otherwise would be desirable or on terms that are less favourable than might otherwise be available or relinquish or license on unfavourable terms our rights to our product candidates in markets where we otherwise would seek to pursue development or commercialization ourselves. If we are unable to obtain sufficient funds in a timely manner, we may be forced to scale back our operating plan; delay or discontinue one or more of our research and development programs; be unable to expand our organization to support our programs; and/or be unable to capitalize on business opportunities as planned. This may negatively impact our business and ability to execute our operating plan.
No assurance can be given that any such additional funding will be available or that, if available, it can be obtained on terms favourable to us. The failure to obtain additional financing on favourable terms, or at all, could have a material adverse effect on our business, financial condition, results of operations and prospects.
- 28 -
It will be many years before the Company expects to generate revenues from product sales to fund its operations, if ever.
The Company does not expect to have a product candidate approved for sale for several years as it advances its technology through the various stages of preclinical development, clinical trial evaluation and regulatory approval process. We have incurred substantial expenses on development efforts to date and have generated operating losses each year since inception. For the years ended December 31, 2025 and 2024, we incurred net losses of US$24.8 million and US$20.6 million respectively and an accumulated deficit of US$81.9 million as at December 31, 2025 and US$57.1 million as at December 31, 2024. We do not expect to generate any revenues from product sales in the immediate future. We may never successfully commercialize any products. Even if we succeed in developing commercial products, we expect to incur additional operating losses for at least the next several years.
As a result, and as is typical for biotechnology companies in such circumstances, the Company anticipates financing its on-going cash requirements for the foreseeable future through a combination of equity offerings, debt financings, government or other third-party funding, marketing and distribution arrangements and other collaborations, partnership agreements and licensing arrangements, and government or philanthropic programs. There are considerable market risks and uncertainties associated with securing such arrangements that may adversely affect the Company’s ability to execute its business plan as intended and negatively impact its stock price.
Risks associated with the Novelty and Early Stage of Development of the Company’s Technologies and Therapeutic Products or Programs
The Company’s programs are subject to significant technical risks and scientific unknowns due to their novelty and stage of development.
The Company’s technology is based on highly original discoveries and hypotheses, and its drug development programs are of an early nature, being in the early clinical development phase. As such, R&D activities over the next 12 months are expected to focus on filing for and obtaining regulatory approvals in numerous jurisdictions to initiate and conduct Phase 2 clinical trials in patients with DMD. This may require additional preclinical development studies to be conducted to support the intended regulatory applications. Technical or scientific setbacks are likely due to the nature of the scientific process and general uncertainties associated with the drug development process. While the Company has anticipated the potential for setbacks in its resource and timeline plans, it is possible that serious unforeseen setbacks could occur in these areas of focus over the next 12 months. Such an eventuality would likely cause the Company to incur additional costs or experience delays in completing its plans on a timely basis, or, depending on their severity, possibly preclude the development and subsequent commercialization of its product candidates. Depending on the gravity of delays or cost increases, the share price of the Company may be adversely affected and its business operations could be materially harmed.
The Company’s drug product candidates may display serious adverse or intolerable side effects during ongoing development.
The Company’s lead drug candidate, SAT-3247, is in the early clinical phase of development. On a statistical basis, drug candidates at this stage of development have an extremely high risk of failure. It is not uncommon for toxicologic, serious adverse or intolerable side effects or lack of sufficient efficacy to be identified during the development of the drug product candidates in the biotechnology industry. If such adverse effects arise, the Company may incur additional costs and/or time delays in attempting to address the underlying issue(s) giving rise to the adverse effects. If the Company is unable to correct the problem on a timely basis or at a reasonable cost, it may need to abandon development of the drug candidate or limit its development to certain uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. Any or all of these outcomes could adversely affect the value of the Company and its stock price, and potentially impair its ability to continue operations.
The Company may not be able to translate its novel discoveries into viable therapeutic treatments suitable for clinical development.
The Company is pioneering a new approach to treating muscle disorders. The Company’s muscle stem cell discoveries are unique and the mechanisms of action it has uncovered have never been modulated for therapeutic purposes in muscle regeneration. The pioneering of this new pharmacology has caused the Company to face certain transaction hurdles. Among other things, the Company will need to determine the appropriate dosing regimens (including dose levels and frequency/interval of dosing) that elicit desirable effects and for how long such effects endure. Determining appropriate dosing regimens may involve significant iterative efforts, take longer than anticipated, or not be achievable at all. Significant delays or a failure to establish the appropriate pharmacological parameters to translate
- 29 -
the Company’s discoveries may severely impede the Company’s product and clinical development progress which may result in materially adverse effects on the Company’s value and stock price, and could impair its ability to continue operations.
Modulating the Notch pathway via AAK1 may reveal safety concerns or require additional safety studies.
Ongoing preclinical and clinical studies will be undertaken by the Company to complement and expand data generated to date for a wide range of drug development reasons. Additionally, preclinical studies will be designed and conducted as required or requested to satisfy regulatory requirements for the purpose of establishing safety tolerances for inhibition of AAK1 with SAT-3247 or SAT-3153 and/or to highlight potential issues that may hinder development for use in humans. While evidence to date suggests to the Company that inhibition of AAK1 at doses anticipated by the Company to be administered in humans to achieve the desired pharmacological effect will have no serious or adverse safety effects, there can be no guarantee that results of the Company’s ongoing or future preclinical safety and toxicological studies will not yield a previously unseen, unknown or unexpected safety concern. Such an outcome could delay expected clinical timelines, introducing additional costs and risk, and may adversely affect the Company’s value and share price. The Company is also conducting toxicology studies at dose levels significantly higher than the anticipated therapeutic dose levels; the results of these studies could also delay expected clinical timelines, introducing additional costs and risk, and may adversely affect the Company’s value and share price.
SAT-3247 or our other development candidates may not be suitable for continuing development in humans.
In addition to safety consideration, preclinical and early clinical studies are often designed to: determine preferred dosing regimens, route of administration (i.e., oral, injection, etc.) and drug levels that yield the desired pharmacological effects prior to testing in humans; establish duration of trials to be conducted in humans; and consider the effects of drug-drug interactions with other drugs prescribed to target patients. The Company is currently conducting such studies, and it is possible that results from such studies may highlight one or more issues which limit the potential utility of SAT-3247 or SAT-3153 as an oral drug candidate for the treatment of Duchenne. While the Company believes that SAT-3247 and SAT-3153 have potential as inhibitors of AAK1 to treat Duchenne patients with an orally available pill or tablet at dose levels which it anticipates could be safe and effective, there can be no guarantee that the results from further preclinical or clinical studies will substantiate this view. Such an outcome could delay or impede clinical trials, introducing additional costs and risk, and may adversely affect the Company’s value and share price.
SAT-3247 may exhibit unexpected issues during ongoing studies which delay, prevent or impede the timely commencement or ongoing conduct of clinical trials.
Prior to commencing clinical trials in humans at each stage of development, successful completion of which are a precondition for ultimately obtaining a product registration and marketing approval, a sponsoring company must submit prescribed dossiers to regulatory authorities in jurisdictions in which it is seeking approval to conduct clinical trials. Typical choices include the FDA in the USA, the EMEA in Europe and Health Canada; the filing dossier is known as either an IND submission in the case of the FDA or a CTA in the case of the EMEA and Health Canada. Prior to submitting an IND/CTA dossier, a body of largely experimental work is undertaken in order to generate the requisite data set expected by the regulators. During the execution of IND-enabling studies, it is possible that untoward effects that had not previously been seen in preclinical studies, such as a dose-limiting toxicity or a manufacturing issue, will manifest. Such findings may introduce material delays or add additional costs to the Company’s timelines and budgets for commencing clinical trials. Additional financing may be required, which may cause dilution to shareholders. Depending on the severity of the issues, the Company’s share price may be negatively affected and the competitive viability of its Duchenne program impaired.
SAT-3247 and our other development candidates may not be patentable.
The Company has made efforts to ensure the novelty of its chemistries and believes SAT-3247 and related compounds are novel inhibitors of AAK1. However, the Company is aware of other inhibitors of AAK1 and it is possible that the Company’s chemical structures could infringe the patents of others. This may not be known for some time. Should this occur, depending on the severity of the possible infringement, the Company’s share price may be negatively affected and the competitive viability of its Duchenne program impaired.
- 30 -
Risks to the Company associated with Clinical and Regulatory Processes
There can be no guarantee that the Company will obtain the required regulatory authorization(s) to commence the Phase 2 clinical trial program on a timely basis, or at all.
Prior to obtaining regulatory approval to register and begin marketing a new drug candidate, a sponsoring company must conduct exhaustive clinical trials in humans, often over many years, to evaluate and determine the safety and efficacy of its drug candidates. Generally, sponsoring companies will seek authorization to conduct clinical trials in humans in one or more jurisdictions such as with the FDA in the USA, the EMEA in Europe or Health Canada once it has completed the requisite preclinical development work. Securing FDA or Health Canada authorization to commence human clinical testing requires the submission of extensive preclinical and supporting information to the FDA or Health Canada for each product candidate to be tested. The process of obtaining authorization to commence human clinical testing, both in the United States and Canada, as well as abroad, is expensive and lengthy and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Regulators such as the FDA or Health Canada have substantial discretion in the authorization process and may refuse to accept any application or may decide that the submitting company’s data is insufficient for approval to commence human clinical testing and/or require additional preclinical or other studies. In addition, varying interpretations of the data obtained from preclinical testing could delay, limit or completely prevent authorization to commence human clinical testing of a product candidate.
Efficacy or other positive results of the Company’s drug candidate(s) seen in preclinical animal model testing may not translate to and be seen in human clinical trials which would have a deleterious effect on the clinical development and potential for regulatory approval of the Company’s drug candidates.
Evaluating drug candidates in human clinical testing can be very costly, challenging to design and implement, and take significant time to complete. These trials are done in a stepped fashion Phases 1 through 3 with the intent to show safety on a continuing, progressive basis through exposure to increased populations of healthy and then patient volunteers while in parallel, exploring and demonstrating the potential for beneficial therapeutic effect. Clinical trials are inherently risky and there can be no assurance that the desired outcomes will be achieved nor that a drug candidate will progress smoothly from one step to the next. It is not uncommon in the biotechnology industry that positive efficacy or other beneficial therapeutic results seen in preclinical testing in animal models of a disease, as has been demonstrated with SAT-3247 and SAT-3153, do not translate in an equivalent manner, or at all, to humans during clinical trial testing as animals are not always predictive of humans. This is true in Duchenne as well, where numerous therapeutic approaches which showed promise in preclinical developments did not demonstrate sufficient promise in human clinical testing to warrant continued development or market approval. There can be no assurance that positive or beneficial results shown by the Company’s drug candidates in preclinical animal model testing will translate to meaningful benefits in humans which are sufficient to justify and support continued clinical development. Such an outcome could potentially lead to the cessation of clinical development of the drug candidate(s) and adversely affect the Company’s market value and possible viability.
Clinical trials in humans may exhibit previously undetected side-effects which may cause additional costs or delays in conducting clinical trials and possibly impede the Company’s ability to complete clinical development of its drug candidate(s).
Unexpected or more severe adverse side effects than seen from preclinical safety testing can occur in human clinical testing as the number of patients treated or duration of treatments increase. There can be no assurance that the Company’s product candidates will not be shown in human clinical testing to have unacceptable side effects or toxicities not seen or predicted from preclinical safety assessments. If clinical trials of SAT-3247 fail to demonstrate adequate safety to the satisfaction of Health Canada and the FDA (or similar regulatory authorities outside the US) on a progressive basis through the phases of clinical development, the Company may incur additional costs or experience delays in completing, or ultimately be unable to complete, the clinical development of its product candidate(s). Such an outcome may severely impair the Company’s value and ability to continue operations.
Clinical trials of SAT-3247 or other development candidates may be protracted due to a limited number of patients available for recruitment and may not be completed on a timely basis.
Duchenne muscular dystrophy is a rare disease, with an estimated total prevalence in the US of 16,000 persons and therefore represents a disease with a small patient pool for clinical testing. This patient pool is further reduced to approximately 2,000 persons when considering the most widely utilized age range of patients between 5 and 9 years which are often enrolled in studies where effects on ambulation are to be evaluated. Although Duchenne patients are often motivated to participate in clinical trials, the small number of patients available, compounded with the growing number of drug candidates entering clinical testing, creates competition for patient
- 31 -
enrollment during clinical studies. Failure to enroll sufficient numbers of patients in a reasonable amount of time in a clinical trial can significantly delay the time to completion of the trial, subsequent outcome analysis of the trial, as well as any future trials that rely on the outcomes of these studies. Such delays or lack of patient enrollment in clinical trials may cause the Company to incur additional costs or experience delays in completing, or ultimately to be unable to complete, the clinical development of its product candidate(s). Such an outcome may severely impair the Company’s value and ability to continue operations.
The FDA has not established a set of required or recommended clinical outcome measurements for studies in Duchenne.
The FDA has produced guidance on the development of drugs for the treatment of Duchenne, however there are no generally accepted criteria for how to design, implement, or evaluate clinical outcome measures that would provide any guarantee of a favourable review for the purpose of drug approval. The FDA has suggested that it will consider the use of existing outcome measures developed for other clinical trials in Duchenne or related muscular dystrophies, as well as proposals for the use of novel outcome measures that are capable of measuring clinically meaningful effects in patients. The FDA encourages sponsors to engage with them early to discuss use of previously utilized measures or the proposition of novel measures prior to submitting official clinical trial designs. The FDA suggests that sponsors should include an assessment of multiple efficacy endpoints, when feasible, to characterize the breadth of effects on dystrophin-related pathologies, including skeletal, respiratory, and cardiac muscle function, even if the primary endpoint is only one of these measures. The open-ended nature of these suggestions may result in selection of inappropriate endpoints that do not yield meaningful results, or a large number of endpoints that significantly increase the cost of clinical trials. Implementing open ended trial designs with multiple endpoints or having to redesign and execute additional trials due to inappropriate selection of clinical endpoints that do not yield meaningful results may cause the Company to incur additional costs or experience delays in completing, or ultimately be unable to complete, the clinical development of its product candidate(s). Such an outcome may impair the Company’s value and ability to continue operations. In addition, as there are not generally accepted biomarkers that have been approved as surrogate endpoints for DMD, and functional outcomes are not necessarily expected to change in a rapid fashion, based on the mechanism of the disease, studies may have to be long in duration to generate positive efficacy data. The functional outcomes in this population are subjective in nature and therefore placebo-controlled study designs will likely be required despite availability of robust natural history data in this orphan indication as multiple prior studies have demonstrated a placebo-arm that behaves differently than natural history data. There is precedence from multiple other sponsors that lead us to assume we will have to conduct placebo-controlled studies, thus increasing the number of patients required for our studies and associated costs and timelines which could have a negative impact on the price of our Common Shares.
Interim, topline and preliminary data from our clinical trials and preclinical studies that we announce or publish from time to time may change as more patient data becomes available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may also disclose interim data from our preclinical studies and clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data becomes available or as patients from our clinical trials continue other treatments for their disease. Adverse differences between preliminary, topline or interim data and final data could significantly harm our business prospects.
From time to time, we may publicly disclose preliminary or topline data from our clinical trials and preclinical studies, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the preliminary or topline results that we report may differ from future results of the same trials, or different conclusions or considerations may qualify such results, once additional data has been received and fully evaluated. Topline and preliminary data also remain subject to audit and verification procedures that may result in the final data being materially different from the data we previously published. As a result, topline and preliminary data should be viewed with caution until the final data is available.
If the Company is unable to enroll DMD patients in clinical trials, it will be unable to complete its clinical trials on a timely basis.
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, the proximity of subjects to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, the ability to obtain and maintain patient consents, the risk that enrolled subjects will drop out before completion, competing clinical trials, and clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications the Company is investigating. Furthermore, the Company relies on
- 32 -
CROs and clinical trial sites to ensure the proper and timely conduct of its clinical trials, and while it has agreements governing their committed activities, the Company has limited influence over their actual performance.
If the Company experiences delays in the completion or termination of any clinical trial of its product candidates or any future product candidates, the development timelines will be delayed, which could increase the overall cost of the clinical development program and reduce the anticipated cash runway of the Company. Any delays in completing clinical trials will increase costs, slow down product candidate development and approval processes and may shorten any periods during which the Company may have the exclusive right to commercialize its product candidates, if approved, all of which may allow the Company’s competitors to bring products to market before it does.
Risks Related to the Company’s Dependence on Third-Party Service Providers
The Company depends on timely access to highly skilled experts to conduct its drug discovery and development activities including in the areas of drug discovery, preclinical drug development, chemistry, manufacturing and controls, developing and submitting regulatory submissions necessary to obtain approval to conduct clinical trials, and conducting clinical trials. Many of these expert skills are quite specialized, expensive to hire and challenging to recruit, particularly at scale. The Company believes its core skills to be in muscle stem cell biology and the professional management of the drug development process. As high-quality expert resources are widely available in the biotechnology industry on a fee-for-service basis through third-party CROs, the Company intends to maintain a lean infrastructure focused on its core capabilities and engage CROs wherever it deems technically advantageous, timely and cost-effective to advance its drug discovery, and preclinical and clinical development activities.
The Company will engage specialized CROs on a fee-for-service basis to conduct its drug discovery and preclinical development including pharmacology and IND-enabling studies; those third parties may not perform satisfactorily, including failing to meet deadlines.
By engaging CROs, the Company is implicitly relying on third-party entities to execute the agreed workplans and deliverables therein to a high skill level, on time and within budget. While the Company intends to use due care in the selection, evaluating and contracting of only the highest quality CROs with reputations for successfully meeting client needs, it is possible that one or more CROs may not meet their contractual obligations on a timely basis or at all. It may not be possible for the Company to effectively redirect their activities or resources to achieve the desired outcomes on time, within budget or to the expected standard, thereby adding further uncertainty. Depending on the severity of the delays, performance deficiencies or cost overruns, the Company may be forced to change to a different CRO or CROs, which can be disruptive. If required to switch or add new CROs, management’s attention and resources may be redirected and constrained until alternative service providers are in place. These changes may have undesirable knock-on effects to other components of the R&D plan and have the potential to extend timelines and increase costs by significant amounts.
The Company will rely on fee-for-service CROs to develop and submit its regulatory filings and to conduct and report on clinical trials for SAT-3247, or other development candidates, and those third parties may not perform satisfactorily, including failing to meet deadlines.
To submit regulatory applications to obtain approval for and to conduct clinical trials in humans for SAT-3247, the Company has engaged third-party service providers with specialized domain expertise for: (i) drafting and submitting IND and CTA submissions and interactions with key regulatory authorities including the FDA and Health Canada; and/or (ii) planning, executing, providing data management and analysis, and reporting of clinical trials with particular expertise in therapies to treat Duchenne. These may and likely will be different entities and could include academic institutions, CROs, hospitals, clinics, and other third-party collaborators. Should the Company be unable to maintain or enter into agreements with these third parties on acceptable terms, or if any such engagement is terminated prematurely, the Company may be unable to enroll patients on a timely basis or otherwise conduct the desired clinical trials in the manner and timeframe originally intended. There is no guarantee that these third parties will devote adequate time and resources to the Company’s clinical studies or perform as required by the agreed terms in the contract or in accordance with regulatory requirements. If these third parties fail to meet expected deadlines, fail to transfer the Company’s regulatory information in a timely manner, fail to adhere to protocols or act in accordance with regulatory requirements, fail to perform in accordance with the agreed contract terms, or if they otherwise perform in a substandard manner or in a way that compromises the quality or accuracy of their activities or the data they obtain, then clinical trials of the Company’s development candidates may be extended or delayed with additional costs incurred, or the clinical trial data may be rejected by the FDA, Health Canada or other regulatory agencies. Such delays, cost increases or rejections could impede or harm SAT-3247’s development and market potential and impair the market value and stock price of the Company.
- 33 -
We rely completely on one third-party contract manufacturer to manufacture SAT-3247 and our other development candidates and we intend to rely on third parties to produce non-clinical, clinical and commercial supplies of SAT-3247 and any other future product candidates.
We do not currently have, nor do we plan to acquire, the infrastructure or capability to internally manufacture our clinical drug supply of SAT-3247, or any other product candidates we may develop in the future, for use in the conduct of our research and development activities, preclinical studies and clinical trials, and we lack the internal resources and the capability to manufacture any product candidates on a clinical or commercial scale.
We plan to continue to rely on contract manufacturers for the foreseeable future to produce quantities of products and substances necessary for research and development, preclinical studies, clinical trials and product commercialization, and to perform their obligations in a timely manner and in accordance with applicable government regulations. While we intend to contract for the commercial manufacture of our product candidates, we may not be able to identify and qualify contractors or obtain favourable contracting terms. If any of the third parties with whom we engage, contract research organizations or other third parties experience shutdowns or other business disruptions, including staffing shortages, production slowdowns or stoppages, or other similar disruptions, our ability to conduct our business in the manner and on the timelines presently planned could be materially and negatively impacted.
Additionally, if our current or future third-party manufacturers do not perform as agreed, experience business disruptions as previously described, or breach or terminate their agreements with us, significant additional time and costs would be required to effect a transition to a new contract manufacturer. If we are unable to retain our current contractors, or are unable to secure arrangements with new contractors to provide manufacturing services in a timely manner and on acceptable terms as needed, it will delay or prevent the development, promotion, marketing, or sale of SAT-3247, if approved, or any other future product candidates we may develop, and have a negative effect on our operations and financial condition. Moreover, if a replacement to our current or future contract manufacturers is required, the ability to establish second-sourcing or find a replacement manufacturer may be difficult due to the lead times generally required to manufacture drug products and the need for regulatory compliance inspections and approvals of any replacement manufacturer, all of which factors could result in production delays and additional costs.
Failure by CRO’s to meet regulatory requirements, guidelines and standards could be injurious to the Company.
The Company and its CROs are required to comply with Good Laboratory Practices (“GLP”) and current Good Clinical Practices (“cGCP”) regulations and guidelines enforced by the FDA, Health Canada, and comparable foreign regulatory authorities. Regulatory authorities enforce these GLP/cGCP regulations through periodic inspections of CRO laboratories, clinical services CROs, principal investigators and clinical trial sites. Failure to comply with applicable cGCP regulations may result in the clinical data generated in clinical trials being deemed unreliable. In addition, clinical trials must typically be conducted with products produced under the cGMP regulations per FDA, Health Canada and other regulatory authorities. Failure to comply with these regulations may impede ongoing clinical development and ultimately, submission of marketing applications may be delayed. Depending on the gravity of the non-compliance, the FDA, Health Canada or other regulatory authorities may demand that clinical trials be repeated, which would cause significant delays and cost increases. If any of the clinical trial sites terminate for any reason, the follow-up information on patients enrolled in an ongoing clinical trial may be lost unless the Company is successfully able to transfer the care of those patients to another qualified clinical trial site, which is not guaranteed. Further, if the Company must terminate the agreements with any of its CROs, it may not be possible to enter into arrangements with alternative CROs on a timely basis or on commercially reasonable terms, or at all. Redoing clinical trials and/or switching CROs during a clinical trial would impose serious pressures on management and could severely delay development and market potential of the Company’s drug candidates, as well as adversely affect its market value and stock price.
Risks Related to Competitive Environment, Drug Development, Challenges, Partnering, Market Access Issues, and Registration and Reimbursement of the Company’s Muscle Regeneration Drug Candidates
There are multiple therapeutic approaches and product candidates currently in clinical development for the treatment of Duchenne including but not limited to gene correction and gene therapy. These or other approaches not yet in clinical development may be approved faster or achieve far better results than anticipated or than the Company’s product candidates.
The Company faces competition from pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies as well as academic institutions, government agencies and other public and private research organizations that conduct research in Duchenne. Many of these competitors or potential competitors have greater financial resources and development, selling and marketing capabilities than the Company. Numerous therapeutic approaches and product candidates including but not limited to: exon skipping,
- 34 -
gene therapy and CRISPR-CAS, anti- fibrosis, myostatin inhibition, mitochondrial and metabolic modulation, and others are in or close to commencing clinical development in humans. While the Company believes its approach is distinctive and may well complement other approaches due to its different mechanism of action, there can be no guarantee that this proves to be the case and one or more competing products may get to market faster with better outcomes for patients, thus reducing the market opportunity for the Company’s product candidates. If the Company’s products are ultimately not competitive, the Company’s revenue and financial results, and general business prospects including market value would be adversely affected.
Satellos may not achieve its publicly announced milestones according to schedule, or at all.
From time to time, Satellos may announce the timing of certain events expected to occur, such as the anticipated timing of initiation of clinical development for SAT-3247 or other therapies or results from clinical trials. These statements are forward-looking and are based on the best estimates of management at the time relating to the occurrence of such events. However, the actual timing of such events may differ from what has been publicly disclosed. The timing of events such as initiation or completion of a clinical trial, filing of an application to obtain regulatory approval or announcement of additional clinical trials for a drug candidate may ultimately vary from what is publicly disclosed. These variations in timing may occur as a result of different events, including the ability to recruit patients in a clinical trial in a timely manner, the nature of results obtained during a clinical trial or during a research phase, problems with a contract manufacturing organization or a contract research organization, or any other event having the effect of delaying the publicly announced timeline. The Company undertakes no obligation to update or revise any forward-looking information, whether as a result of new information, future events or otherwise, except as otherwise required by law. Any variation in the timing of previously announced milestones could have a material adverse effect on the business plan, financial condition or operating results of the Company and the trading price of the Common Shares.
If sufficient beneficial advantages for patients are not shown in clinical trials, the Company may be unable to secure development partners on favourable terms, or at all.
The Company may pursue development and commercialization partnership arrangements with third parties. Potential partners for any clinical development, co-marketing, licensing or broader arrangements may include large and mid-size pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies. Often partnership agreements are made to obtain up-front capital, offset development costs, increase the probability of success from leveraging the skill sets of a more experienced partner, and secure a royalty annuity. In return the partner generally receives downstream commercialization rights (or co-rights) on a region-by-region basis, or globally. The Company may benefit from such a partnership particularly given the pediatric nature of Duchenne, complexity of disease progression and its sequalae, and the specialization of the medical centers which treat patients. Failure to demonstrate sufficient patient benefit may make it more difficult or preclude the Company from being successful in establishing these agreements should it choose to do so. This may increase the Company’s business costs and risks, delay commercialization activities and revenue generation, and potentially impair the business.
Despite launching products in the future, the Company may experience limits in market access, unfavourable reimbursement or pricing, or healthcare policy reform initiatives, which would limit the value of the products.
The Company’s ability to commercialize any products successfully will depend, in part, on the extent to which coverage and reimbursement for these products and related treatments will be available from government healthcare programs, private health insurers, managed care plans, and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels. A primary trend in the United States healthcare industry is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. The Company cannot be sure that coverage and reimbursement will be available on a timely basis for any product that it, or any of its partners, commercializes and, if reimbursement is available, the level of reimbursement may be reduced. Access to reimbursement may impact the demand for, or the price of, any product candidate for which the Company or its partner(s) obtains marketing approval. If reimbursement is not available on a timely basis or is available only to limited levels or is delayed, the commercial potential for any product candidate for which the Company or its partner has obtained marketing approval may be reduced. Such an outcome could adversely affect the Company’s profitability and share price.
- 35 -
Product liability lawsuits could cause reputational damage and legal liabilities for the Company, limiting the commercial prospects for its products.
There is an inherent risk of product liability exposure related to the future evaluation of the Company’s product candidates in human clinical trials. The Company’s current product candidates have not been tested in human clinical trials, and therefore, safety data is limited and the data which has been generated to date may not translate to humans. If the Company cannot successfully defend itself against claims that its product candidates or products caused injuries, it will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in decreased demand for any product candidates or products that the Company may develop, reputational injury and negative attention, withdrawal of clinical trial participants, significant costs to defend the related litigation, potential monetary awards to trial participants or patients, and lost or delayed product revenues. Such outcomes would be injurious to the Company’s business.
Liability Insurance may not be adequate.
The Company does not yet hold clinical trial liability insurance coverage, and any coverage that may be obtained in the future may not be adequate to cover all liabilities that it may incur. When the Company begins to commercialize its product candidates, it will need to increase its insurance coverage, which is increasingly expensive. The Company may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
Risks Related to the Company’s Intellectual Property and Privacy Legislation
If the Company fails to comply with its obligations under its intellectual property licenses with third parties, it could lose license rights that are important to its business.
The Company is party to intellectual property license agreements with the OHRI and expects to enter into additional license agreements in the future. The Company’s existing license agreements impose, and future license agreements will most likely impose, various milestone payments, royalties, insurance, indemnification and other obligations on the Company.
The Company’s current agreement with the OHRI requires the Company to maintain its patents and pay annual license fees and research fees. If the Company fails to comply with its obligations under this license, the OHRI may have the right to terminate this license agreement. In such event, the Company might not be able to market any product that is covered by such license, or to convert such license to a non-exclusive license. This could materially adversely affect the value of the product candidate being developed under the OHRI License. Termination of any license agreement or reduction or elimination of the Company’s licensed rights may result in the Company having to negotiate new or reinstated licenses with less favourable terms.
If the Company is unable to obtain and maintain patent protection for its technology and products, or if the Company’s partners or licensors are unable to obtain and maintain patent protection for the technology or products that it licenses from them, or if the scope of the patent protection obtained is not sufficiently broad, the Company’s competitors could develop and commercialize technology and products similar or identical to that of the Company, and its ability to successfully commercialize its technology and products may be adversely affected.
The Company’s success will depend on its ability to obtain and maintain patent and other intellectual property protection with respect to its product candidates. The Company and its licensors have sought to protect the Company’s proprietary position by filing patent applications in the United States, Canada, and abroad related to its novel technologies and products that are important to its business. This process is expensive and time-consuming, and the Company may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. In addition, patents might not be issued or granted with respect to the Company’s patent applications that are currently pending, and issued or granted patents might later be found to be invalid or unenforceable, be interpreted in a manner that does not adequately protect the Company’s current product or any future products or fail to otherwise provide us with any competitive advantage. The patent position of biotechnology and pharmaceutical companies is generally uncertain because it involves complex legal and factual considerations and in recent years has been the subject of much litigation. The standards applied by the United States Patent and Trademark Office (the “USPTO”) and foreign patent offices in granting patents are not always applied uniformly or predictably. As a result, the issuance, scope, validity, enforceability and commercial value of the Company’s and its partners’ or licensors’ patent rights are highly uncertain. The degree of future protection that the Company will have on its proprietary products and technology, if any, is uncertain and a failure to obtain adequate intellectual property protection with respect to the Company’s product candidates and proprietary technology could have a material adverse impact on the success of its business.
- 36 -
Even if the Company’s owned and licensed patent applications issue as patents, they may not issue in a form that will provide the Company with any meaningful protection, prevent competitors from competing with the Company or otherwise provide the Company with any competitive advantage, including not being listed in the FDA’s Approved Drug Products With Therapeutic Equivalence Evaluations publication (commonly known as, the Orange Book). The Company’s competitors may be able to circumvent its owned or licensed patents by developing similar or alternative technologies or products in a non-infringing manner. The issuance of a patent is not conclusive as to its scope, validity or enforceability, and the Company’s owned and licensed patents may be challenged in the courts or patent offices in Canada, the United States and abroad. Such challenges may result in patent claims being narrowed, invalidated or held unenforceable, which could limit the Company’s ability to or stop or prevent the Company from stopping others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of its technology and products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, the Company’s owned and licensed patent portfolio may not provide it with sufficient rights to exclude others from commercializing products similar or identical to the Company’s.
The Company may become involved in lawsuits to protect or enforce its patents, which could be expensive, time consuming and, whether successful or unsuccessful, limit the commercial value of the Company’s product or have a material adverse effect on the Company’s business.
Competitors may infringe any of the Company’s current or future patents. To counter infringement or unauthorized use, the Company may be required to file expensive and time-consuming infringement claims. Also, the court may decide in an infringement proceeding that a specific patent held by the Company is not valid or enforceable or may refuse to stop the other party from using the Company’s intellectual property on the grounds that its patents do not cover the intellectual property being disputed. An adverse result in any litigation proceeding could put one or more of the Company’s patents at risk of being invalidated or interpreted narrowly. Additionally, due to the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of the Company’s confidential information could be compromised by disclosure during this type of litigation. In addition, the Company’s licensor may have rights to file and prosecute such claims and it is reliant on them.
The Company’s commercial successes depend upon its ability and the ability of its partners and other collaborators to develop, manufacture, market and sell its product candidates and use its proprietary technologies without infringing the proprietary rights of third parties. Third parties may assert infringement claims against the Company based on existing patents or patents that may be granted in the future which at this time cannot be known to the Company. The Company may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to its products and technology, including interference proceedings before the USPTO or other similar regulatory authorities. If the third party is successful and the Company is found to infringe on their intellectual property rights, the Company could be forced to negotiate the rights to the third party’s intellectual property in order to continue to develop and market the Company’s products and technology. There is no guarantee that the Company will be able to obtain any required license on commercially reasonable terms or at all. Even if the Company was able to obtain a license, it could be non-exclusive, thereby giving its competitors access to the same technologies licensed to the Company. If the Company is not able to obtain a license for the rights to their technology, the Company could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, the Company could be found liable for additional monetary damages. A finding of infringement could prevent the Company from commercializing its product candidates, or delay commercialization during adjudication of a patent dispute, including a 30-month injunction, or force the Company to cease some of its business operations, pay royalties and/or damages to companies holding the patents that were infringed, all of which could materially harm the Company’s business. Claims that the Company has misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on its business.
Litigation or other legal proceedings relating to intellectual property claims may cause the Company to incur significant expenses and could distract the Company’s employees from their normal responsibilities, even if it is resolved in the Company’s favour. Also, any public announcements of the results of hearings, motions or other interim proceedings or developments could be perceived to be negative by securities analysts or investors, leading to a potential adverse effect on the price of the Common Shares. These types of litigation or proceedings could substantially increase the Company’s operating losses and reduce the resources available for product development activities. The Company may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of the Company’s competitors may be able to sustain the costs of such litigation or proceedings more effectively than it can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on the Company’s ability to compete in the marketplace.
- 37 -
The Company may be subject to claims that its employees have wrongfully used or disclosed alleged trade secrets of their former employers.
The Company makes efforts to ensure that its employees do not use the proprietary information or know-how of others in their work for the Company; however, the Company may nonetheless be subject to claims that it or these employees have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer. This would most likely result in the Company having to enter litigation to defend against these claims. If the Company fails in defending any such claims, in addition to paying monetary damages, it may lose valuable intellectual property rights and/or personnel. Even if the Company is successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
If the Company is unable to protect the confidentiality of its trade secrets, the Company’s innovative capacity and competitive position could be harmed.
The Company relies on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain its competitive position, in addition to filing patents for some of its technology and products. The types of protections available for trade secrets are particularly important with respect to the Company’s proprietary MyoRegenXTM technology platform, which involves significant unpatented know-how. The Company seeks to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as the Company’s employees, corporate collaborators, outside scientific collaborators, sponsored researchers, contract manufacturers, consultants, advisors and other third parties. The Company also enters into confidentiality and invention or patent assignment agreements with its employees and consultants. Despite these efforts, any of these parties may breach the agreements and disclose the Company’s proprietary information, including its trade secrets, and the Company may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, courts in certain jurisdictions are less willing or unwilling to protect trade secrets. If any of the Company’s trade secrets were to be lawfully obtained or independently developed by a competitor, it would have no right to prevent them from using that technology or information to compete with the Company. If any of the Company’s trade secrets were to be disclosed to or independently developed by a competitor, its competitive position would be harmed.
The Company must protect and manage confidential personal health information, including reporting from marketed product adverse event reporting and clinical trials. Accidental release of information could harm the Company.
As the Company’s programs advance in development, the Company expects to generate or otherwise obtain clinical data that may include personal information and personal health information. This data is required for successful development and commercialization of pharmaceutical products, such as clinical trial data to support regulatory submissions and pharmacovigilance data to monitor for potential adverse events following product launch. The Company recognizes the sensitivity of this data and will apply protections to minimize the risk of release, including strict data blinding protocols and secure information technology infrastructure. However, despite these measures, it is possible that personal information or personal health information could be released and may expose the Company to substantial reputational risk and legal liabilities. Regardless of merit or eventual outcome, liability claims may result in decreased demand for any product candidates or products that it may develop, injury to the Company’s reputation and significant negative media attention, withdrawal of clinical trial participants, significant costs to defend the related litigation, substantial monetary awards to trial participants or patients, loss of revenue and the inability to commercialize any products that the Company may develop.
We rely on patents and other intellectual property rights to protect our product candidates, the enforcement, defense and maintenance of which may be challenging and costly. Failure to enforce or protect these rights adequately could harm our ability to compete and impair our business.
Our commercial success depends in part on obtaining and maintaining patents and other forms of intellectual property rights for our product candidates, including SAT-3247, any future therapeutic candidates, any methods used to manufacture the underlying drug substances, and the methods for treating patients using those substances, or on licensing in such rights. Failure to obtain, maintain, protect, enforce or extend adequate patent and other intellectual property rights could materially adversely affect our ability to develop and market our product candidates and any future therapeutic candidates. We also rely on trade secrets and know-how to develop and maintain our proprietary and intellectual property position. Any failure to protect our trade secrets and know-how could adversely affect our operations and prospects.
- 38 -
We cannot be certain that patents will be issued or granted with respect to patent applications that are currently pending, or that issued or granted patents will not later be found to be invalid or unenforceable. The patent position of companies like ours is generally uncertain because it involves complex legal and factual considerations. The standards applied by the European Patent Office, the USPTO, and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in pharmaceutical patents. Consequently, patents may not issue from our pending patent applications, and even if they do issue, such patents may not issue in a form that effectively prevents others from developing or commercializing competing treatments. As such, we do not know the degree of future protection that we will have on our proprietary treatments.
The patent prosecution process is expensive, complex and time-consuming, and we and our current or future third-party partners, licensors, licensees, or collaboration partners may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we or our licensors, licensees or collaboration partners will fail to identify patentable aspects of inventions made in the course of research, development or commercialization activities before it is too late to pursue patent protection on them. In addition, although we enter into non-disclosure and confidentiality agreements with parties who have access to confidential or patentable aspects of our research and development output, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors, and other third parties, any of these parties may breach the agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection. Furthermore, publications of discoveries in scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not published until and unless granted. Therefore, we cannot be certain that we were the first to make the inventions claimed in our patents or pending patent applications, or that we were the first to file for patent protection of such inventions. Similarly, we cannot be certain that for any licensed patents or pending patent applications, the named applicant(s) were the first to make the inventions claimed in such patents or pending patent applications or that the named applicant(s) were the first to file for patent protection for such inventions.
Further, the issuance, scope, validity, enforceability and commercial value of our and our current or future licensors’, licensees’ or collaboration partners’ patent rights are highly uncertain. Our and our licensors’ pending and future patent applications may not result in patents being issued that protect our treatments, in whole or in part, or that effectively prevent others from commercializing competitive technologies and treatments.
Moreover, in some circumstances, we may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, covering technology that we license from or license to third parties and are reliant on our licensors, licensees or collaboration partners. If our current or future licensors, licensees or collaboration partners fail to establish, maintain or protect such patents and other intellectual property rights, such rights may be reduced or eliminated. If our licensors, licensees or collaboration partners are not fully cooperative or disagree with us as to the prosecution, maintenance or enforcement of any patent rights, such patent rights could be compromised.
The patent examination process may require us or our licensors, licensees or collaboration partners to narrow the scope of the claims of our or our licensors’, licensees’ or collaboration partners’ pending and future patent applications, which may limit the scope of patent protection that may be obtained. We cannot assure you that all of the potentially relevant prior art relating to our patents and patent applications has been found. If such prior art exists, it can invalidate a patent or prevent a patent from issuing from a pending patent application.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the United States and abroad. Even if patents do successfully issue and even if such patents cover our product candidates and any future therapeutic candidates, third parties may initiate an opposition, interference, re-examination, post-grant review, inter partes review, nullification or derivation proceedings in court or before patent offices, or similar proceedings challenging the validity, enforceability or scope of such patents, which may result in the patent claims being narrowed or invalidated.
We may be forced to litigate to enforce or defend our intellectual property rights.
We may be forced to litigate to enforce or defend our intellectual property rights against infringement by competitors, and to protect our know-how against unauthorized use, but we may not be able to detect or prevent, alone or with our licensors, infringement, misappropriation or other violation of our intellectual property rights. In so doing, we may place our intellectual property at risk of being invalidated, rendered unenforceable or limited or narrowed in scope such that we may no longer be able to adequately prevent the manufacture, sale or import of competitive product. In an infringement proceeding, a court may decide that a patent we own or a patent
- 39 -
we may license in the future is invalid or unenforceable or may refuse to stop the other party from using the invention at issue. Even if we establish infringement, the court may decide not to grant an injunction against further infringing activity and instead award only monetary damages, which may or may not be an adequate remedy. Further, an adverse result in any litigation or other proceedings before government agencies such as the USPTO, may place pending applications at risk of non-issuance or limitations in scope. Further, derivation proceedings, ex parte reexamination, inter partes review, post grant review and opposition proceedings provoked by third parties or brought by the USPTO or any foreign patent authority may be used to challenge the inventorship, ownership, claim scope or validity of our patents. Additionally, because of the substantial amount of discovery typically required in connection with intellectual property litigation, there is a risk that some of our confidential and proprietary information or know-how could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the value of the company. Such litigation or proceedings could substantially increase our operating losses, reduce the resources available for development activities or any future sales, marketing or distribution activities and distract our personnel from their normal responsibilities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
If we become involved in patent litigation or other proceedings related to a determination of rights, we could incur substantial costs and expenses, substantial liability for damages or be required to stop our product development and commercialization efforts.
Our commercial success depends in part on our ability and the ability of any of our future partners to develop, manufacture, market and sell our product candidates and use our proprietary technologies without infringing on the intellectual proprietary rights of third parties. There is a substantial amount of litigation and patent office proceedings, both within and outside the United States, involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, oppositions, ex parte reexaminations, post-grant review, inter partes review and interference proceedings before the USPTO and corresponding foreign patent offices. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are pursuing product candidates.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims to compositions, formulations, methods of manufacture or methods for treatment relating to our product candidates, their manufacture or use. Because patent applications can take many years to issue, there may be pending patent applications which may later result in issued patents that our product candidates may be alleged to infringe. In addition, third parties may obtain patents in the future and then claim that our technologies infringe upon these patents. We cannot guarantee that any of our or our licensors’ patent searches or analyses, including the identification of relevant patents, the scope of patent claims or the expiration of relevant patents, are or will be complete or thorough, nor can we be certain that we or our licensors have identified or will identify each and every third party patent and pending patent application in the United States and abroad that is relevant to or necessary for the commercialization of our current and future products and product candidates in any jurisdiction. Our interpretation of the relevance or the scope of a patent or a pending patent application may be incorrect, which may negatively impact our ability to market our products. We may incorrectly determine that our products or product candidates are not covered by a third-party patent or may incorrectly predict whether a third party’s pending patent application will issue with claims of relevant scope. Alternatively, we may incorrectly determine that the Hatch-Waxman Amendments (as defined below) are a defense for a safe harbour to infringement of a patent we consider relevant to the research or clinical development of our product candidates. Our determination of the expiration date of any patent in the United States or abroad that we consider relevant may be incorrect, and we may incorrectly conclude that a third-party patent is invalid and unenforceable or not infringed. Our failure to identify and correctly interpret relevant patents may negatively impact our ability to develop and market our products and product candidates. If we fail to identify and correctly interpret relevant patents, we may be subject to infringement claims. As the number of competitors in the market grows and the number of patents issued in this area increases, the possibility of patent infringement claims escalates. Defense of infringement and other claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business.
In such cases, we may not be in a position to develop or commercialize such product candidates unless we successfully pursue litigation to invalidate the third-party intellectual property right concerned, or enter into a license agreement with the intellectual property right holder, which may not be available on commercially reasonable terms or at all. In the event that a patent has not expired at the time of approval of such product candidate and the patent owner were to bring an infringement action against us, we may have to argue that our product candidates or the manufacture or use of the underlying therapeutic substances do not infringe a valid claim of the patent in
- 40 -
question. Alternatively, if we were to challenge the validity of any issued U.S. patent in court, we would need to overcome a statutory presumption of validity that attaches to every U.S. patent. This means that in order to prevail, we would need to present clear and convincing evidence as to the invalidity of the patent’s claims. The same applies to other jurisdictions. Even if we believe third-party intellectual property claims are without merit, there is no assurance that a court would find in our favour on questions of infringement, validity, enforceability, or priority. In the event that a third party successfully asserts its patent against us such that such third party’s patent is found to be valid and enforceable and infringed by our product candidates, unless we obtain a license to such patent, which may not be available on commercially reasonable terms or at all, we could be prevented from continuing to commercialize our product candidates.
If we are unsuccessful defending a claim of infringement and obtaining a license, in addition to being forced to pay damages, we may be temporarily or permanently prohibited from commercializing any of our product candidates that were held to be infringing. If possible, we might be forced to redesign our product candidates so that we no longer infringe the intellectual property rights of third parties. Even if we or our licensors or collaboration partners obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us or our licensors or collaboration partners and it could require us to make significant licensing and royalty payments. In addition, we could be found liable for significant monetary damages, including treble damages and attorneys’ fees, if we are found to have willfully infringed a patent or other intellectual property right. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar material adverse effect on our business, financial condition, results of operations, and prospects. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
Our rights to develop and commercialize our product candidates are, and in the future may be, subject to the terms and conditions of licenses granted to us by others. If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties, or these agreements are terminated, or we otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business.
We are dependent on patent rights, know-how and proprietary technology licensed from third parties, including through a license agreement with the OHRI. We may also enter into additional license agreements with third parties in the future. Our current and future license agreements may impose diligence, development and commercialization timelines, milestone payments, royalties, indemnification, insurance or other obligations on us. If we fail to comply with our obligations to our licensors or collaborators, our counterparties may have the right to terminate these agreements. Termination of these agreements or reduction or elimination of our rights under these agreements may result in us having to negotiate new or reinstated agreements with less favourable terms or cause us to lose our rights under these agreements, including our rights to important intellectual property or technology that are necessary for our business.
Our success will depend in part on the ability of our licensors to obtain, maintain and enforce patent protection for our licensed intellectual property. We may have limited control over our licensors’ activities or use or licensing of any other intellectual property that may be related to our in-licensed intellectual property. Our licensors may not successfully prosecute the patent applications we license. Even if patents issue from these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than we would. If any of our licensors or licensees having rights to file, prosecute, maintain and defend our patent rights fail to conduct these activities for patents or patent applications covering any of our product candidates, our ability to develop and commercialize those product candidates may be adversely affected and we may not be able to prevent competitors or other third parties from making, using or selling competing products. In addition, we may sublicense certain of our rights under various third-party licenses to our strategic collaborators. Any impairment of these sublicensed rights could result in reduced revenues under our strategic collaboration agreements or result in termination of an agreement by one or more of our strategic collaborators. In addition, intellectual property rights that we may in-license in the future may be sublicensed under intellectual property owned by third parties, in some cases through multiple tiers. The actions of our licensors may therefore affect our rights to use our sublicensed intellectual property, even if we are in compliance with all of the obligations under our license agreements. Should our licensors or any of the upstream licensors fail to comply with their obligations under the agreements pursuant to which they obtain the rights that are sublicensed to us, or should such agreements be terminated or amended, our ability to develop and commercialize our product candidates may be materially harmed.
We cannot be certain that such activities by our licensors have been or will be conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents or other intellectual property rights. Pursuant to the terms of the license agreements with our licensors, such licensors may have the right to control enforcement of our licensed patents or defense of any claims asserting the invalidity of such patents and, even if we are permitted to pursue such enforcement or defense, we cannot ensure the
- 41 -
cooperation of our licensors or, in some cases, other necessary parties, such as any co-owners of patents or other intellectual property from which we have not yet obtained a license. We cannot be certain that our licensors will allocate sufficient resources or prioritize their or our enforcement of such patents or defense of such claims to protect our interests in the licensed patents. Even if we are not a party to these legal actions, an adverse outcome could harm our business because it might prevent us from continuing to license intellectual property that we may need to operate our business. In addition, even when we have the right to control patent prosecution of licensed patents and patent applications, enforcement of licensed patents, or defense of claims asserting the invalidity of those patents, we may still be adversely affected or prejudiced by actions or inactions of our licensors and their counsel that took place prior to or after assuming control.
Our current or future license agreements may not provide exclusive or sufficient rights to use such intellectual property and technology in all relevant fields of use and in all territories in which we may wish to develop or commercialize our product candidates in the future. Some licenses granted to us may be subject to certain preexisting rights held by the licensors or certain third parties. As a result, we may not be able to prevent third parties from developing and commercializing competitive products in certain territories or fields.
In the event that our third-party licensors determine that, in spite of our efforts, we have materially breached a license agreement or have failed to meet certain obligations thereunder, it may elect to terminate the license agreement or, in some cases, one or more license(s) under the applicable license agreement. Such termination could result in us losing the ability to develop and commercialize product candidates and technology covered by the licensed intellectual property. In the event of such termination of a third-party in-license, or if the underlying patent rights under a third-party in-license fail to provide the intended exclusivity, third parties may be able to seek regulatory approval of, and to market, products identical to ours and we may be required to cease the development and commercialization of our product candidates. Moreover, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights. Any of these events could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
In addition, the agreements under which we license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant patents, know-how and proprietary technology, or increase what we believe to be our financial or other obligations under the relevant agreement. Disputes may also arise between us and our licensors regarding intellectual property subject to a license agreement, including: the scope of rights granted under the license agreement and other interpretation-related issues; whether and the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; our right to sublicense patent and other rights to third parties under collaborative development relationships; our diligence obligations with respect to the use of the licensed technology in relation to our development and commercialization of our product candidates, and what activities satisfy those diligence obligations; and the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our strategic collaborators.
If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on favourable terms, we may be unable to successfully develop and commercialize the affected product candidates.
We are generally also subject to all of the same risks with respect to protection of intellectual property that we license, as we are for intellectual property that we own, which are described below. If we or our licensors fail to adequately protect this intellectual property, our ability to commercialize products could suffer.
Any of our future collaboration arrangements may not be successful, which could adversely affect our ability to develop and commercialize our products.
We may seek to collaborate on the development or commercialization of our product candidates. Any of our future collaborations may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborations are subject to numerous risks, which may include that:
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates; |
| ● | collaborators may not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on trial or test results, changes in their strategic focus due to the |
- 42 -
| acquisition of competitive products, availability of funding, or other external factors, such as a business combination that diverts resources or creates competing priorities; |
| ● | disputes may arise between us and a collaborator that causes the delay or termination of the research, development or commercialization of our current or future products or that results in costly litigation or arbitration that diverts management attention and resources; |
| ● | collaborators have significant discretion in determining the efforts and resources that they will apply to collaborations; |
| ● | a collaborator with marketing, manufacturing and distribution rights to one or more products may not commit sufficient resources to or otherwise not perform satisfactorily in carrying out these activities; |
| ● | we could grant exclusive rights to our collaborators that would prevent us from collaborating with others; |
| ● | collaborators may use our intellectual property or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential liability or business risk; |
| ● | collaborations may be terminated, and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable current or future products or products; and |
| ● | collaborators may own or co-own intellectual property covering our product candidates that results from our collaborating with them, and in such cases, we would not have the exclusive right to develop or commercialize such intellectual property; and a collaborator’s sales and marketing activities or other operations may not be in compliance with applicable laws resulting in civil or criminal proceedings. |
We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
We may be subject to claims that former employees, collaborators or other third parties have an ownership interest in our patents or other intellectual property. Ownership disputes may arise, for example, from conflicting obligations of consultants or others who are involved in developing our product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse impact on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
In addition, while it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own. The assignment of intellectual property rights may not be self-executing, or the assignment agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they may bring against us, to determine the ownership of what we regard as our intellectual property. Such claims could have a material adverse effect on our business, financial condition, results of operations and prospects.
If we enter into future arrangements involving government funding, and we make inventions as a result of such funding, our intellectual property rights to such discoveries may be subject to the applicable provisions of the Bayh Dole Act of 1980.
If we enter into future arrangements involving government funding, and we make inventions as a result of such funding, our intellectual property rights to such discoveries may be subject to the applicable provisions of the Bayh Dole Act of 1980 (the “Bayh Dole Act”). To the extent any of our current and future intellectual property is generated through the use of U.S. government funding, the provisions of the Bayh Dole Act may similarly apply. Any exercise by the government of certain of its rights could harm our competitive position, business, financial condition, results of operations and growth prospects.
U.S. government rights in certain inventions developed under a government-funded program include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for governmental purposes. In addition, the U.S. government would have the right to require us to grant exclusive, partially exclusive or non-exclusive licenses to any of these inventions to a third party if the government determines that: (i) adequate steps have not been taken to commercialize the invention; (ii) government action is necessary to meet public health or safety needs; or (iii) government action is necessary to meet requirements for public use under federal regulations, which we refer to as march-in rights. The U.S. government would also have the right to take title to these inventions if we fail, or the applicable licensor fails, to disclose the invention to the government, elect title and file an application to register the intellectual property within specified time limits. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us, or the applicable licensor, to expend substantial resources. In addition, the U.S.
- 43 -
government requires that any products embodying the subject invention or produced through the use of the subject invention be manufactured substantially in the United States. The manufacturing preference requirement can be waived if the owner of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the United States or that under the circumstances domestic manufacture is not commercially feasible.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents or applications will be due to be paid to the USPTO and various non-U.S. patent agencies in several stages over the lifetime of the patents or applications. The USPTO and non-U.S. patent agencies also require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse impact on our business.
In addition, public health pandemics, geopolitical instability, natural disasters, or similar events may impair our and our licensors’ ability to comply with these procedural, document submission, fee payment, and other requirements imposed by government patent agencies, which may materially and adversely affect our ability to obtain or maintain patent protection for our product candidates. There could also be delays at the USPTO caused by staffing cuts and other U.S. government actions as a result of the U.S. Department of Government Efficiency or other executive actions to reduce the size of the U.S. government.
The USPTO and various non-U.S. government agencies require compliance with certain foreign filing requirements during the patent application process. For example, in some countries, including the United States, China, India and some European countries, a foreign filing license is required before certain patent applications are filed. The foreign filing license requirements vary by country and depend on various factors, including where the inventive activity occurred, citizenship status of the inventors, the residency of the inventors and the invention owner, the place of business for the invention owner and the nature of the subject matter to be disclosed (e.g., items related to national security or national defense). In some, but not all cases, for example in China and India, a foreign filing license cannot be obtained retroactively in accordance with the applicable rules. There are situations, however, in which non-compliance can result in abandonment of a pending patent application or can be grounds for revoking or invalidating an issued patent, resulting in the loss of patent rights in the relevant jurisdiction. In such an event, potential competitors might be able to enter the relevant markets with similar or identical products or technology, which could have a material adverse effect on our business, financial condition, results of operations and prospects. We may also be dependent on our licensors to take the necessary actions to comply with these requirements with respect to our licensed intellectual property.
If we do not obtain sufficient patent term for our product candidates, our business may be materially harmed.
Patents have a limited term. The terms of individual patents depend upon the legal term for patents in the countries in which they are granted. In most countries, including the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from the earliest non-provisional filing date in the applicable country. However, the actual protection afforded by a patent varies from country to country, and also depends upon many factors, including the type of patent, the scope of coverage, the availability of regulatory related extensions, the availability of extensions for patent office delays during the examination process, the availability of legal remedies in a particular country and the validity and enforceability of the patent, and whether a portion of the patent term has been terminally disclaimed based on other patents. Various extensions including patent term extension and patent term adjustment may be available, but the lives of such extensions, and the protections they afford, are limited in the United States. Even if patents covering our product candidates are obtained, once the patent life has expired for a product candidate, we may be open to competition from generics or biosimilars.
Depending upon the timing, duration and specifics of FDA regulatory approval of our product candidates, one or more patents issued from U.S. patent applications that we or a future licensor files may be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984 (the “Hatch-Waxman Amendments”). The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during the FDA regulatory review process based on the
- 44 -
first regulatory approval for a particular drug or biologic. A maximum of one patent may be extended per FDA-approved drug as compensation for the patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of drug approval, and only those claims covering such approved drug product, a method for using it or a method for manufacturing it may be extended. Patent term extension may also be available in certain foreign countries upon regulatory approval of our product candidates.
Despite the possibility of an extension, we may not be granted an extension in the United States or another jurisdiction because of, for example, failure to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time or the scope of patent protection afforded could be less than we request.
If we are unable to obtain patent term extension or restoration, or the foreign equivalent, or the term of any such extension is less than we request, our competitors or other third parties may obtain approval of competing drugs following our patent expiration, and our revenue could be reduced, possibly materially. Further, if this occurs, our competitors or other third parties may take advantage of our investment in development and trials by referencing our clinical and preclinical data and launch their drug earlier than might otherwise be the case. Any of the foregoing could materially harm our business, financial condition, results of operations and growth prospects.
We may enjoy only limited geographical protection with respect to certain patents and we may not be able to protect our intellectual property rights throughout the world.
Filing and prosecuting patent applications and defending patents covering our product candidates in all countries throughout the world would be prohibitively expensive. Competitors may use our technologies and innovations in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but enforcement rights are not as strong as those in the United States, Europe or Canada. These products may compete with our product candidates, and our and our licensors’ future patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
In addition, we may decide to abandon national and regional patent applications before they are granted. The examination of each national or regional patent application is an independent proceeding. As a result, patent applications in the same family may issue as patents in some jurisdictions, such as in the United States, but may issue as patents with claims of different scope or may even be refused in other jurisdictions. Furthermore, the requirements for patentability differ in certain jurisdictions and countries. Some countries do not grant claims directed to methods of treatment or have additional restrictions on the scope of method of treatment claims compared to the United States. Accordingly, depending on the country, the scope of patent protection may vary for the same product candidate.
While we intend to protect our intellectual property rights in our expected significant markets, we cannot ensure that we will be able to initiate or maintain protection efforts in all such markets. Additionally, the prosecution of patent applications in other jurisdictions is often a longer process and patents may be granted at a later date than in the United States, potentially delaying our ability to assert such patents against competitors. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate, which may have an adverse effect on our ability to successfully commercialize our product candidates in all of our expected significant foreign markets. If we encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished, and we may face additional competition in those jurisdictions.
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws or rules and regulations in the United States, Europe and Canada, and many companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favour the enforcement of patents, trade secrets and other intellectual property rights, which could make it difficult for us to stop the infringement of any patents we obtain or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in other jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business, could put any patents we obtain at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing as patents, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
- 45 -
Some countries also have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, some countries limit the enforceability of patents against government agencies or government contractors. In those countries, the patent owner may have limited remedies, which could materially diminish the value of such patents. If we are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired.
In Europe, a new unitary patent system took effect on June 1, 2023, which may significantly impact European patents, including those granted before the introduction of the new system. Under the new system, applicants can, upon grant of a European patent, opt for that patent to become a unitary patent which will be subject to the jurisdiction of a new unitary patent court (“UPC”). During the first seven years of the UPC’s existence, patents granted before the implementation of the new system can be opted out of UPC jurisdiction, and validated as national patents in any one or more of the UPC countries. We may decide to opt out our future European patents from the UPC, but doing so may preclude us from realizing the benefits of the UPC. Moreover, if we do not meet all of the formalities and requirements for opt-out under the UPC, our future European patents could remain under the jurisdiction of the UPC. Patents that are under the jurisdiction of the UPC may be challenged in a single UPC-based revocation proceeding that, if successful, could invalidate the patent in all countries who are signatories to the UPC. The UPC will provide our competitors with a new forum to centrally revoke our European patents, and allow for the possibility of a competitor to obtain pan-European injunction. Further, because the UPC is a relatively new court system and there is little precedent for the court’s laws, there is increased uncertainty regarding the outcome of any patent litigation. We are unable to predict what impact the new patent regime may have on our ability to exclude competitors in the European market. In addition to changes in patents laws, geopolitical dynamics, such as Russia’s incursion into Ukraine, may also impact our ability to obtain and enforce patents in particular jurisdictions, such as the enforcement of patent rights in Russia. If we are unable to obtain and enforce patents as needed in particular markets, our ability to exclude competitors in those markets may be reduced.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining, defending, maintaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity and is therefore costly, time consuming and inherently uncertain. Changes in either the patent laws or interpretation of the patent laws in the United States and in other major jurisdictions could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents, and may diminish our ability to protect our inventions, obtain, maintain, enforce and protect our intellectual property rights and, more generally, could affect the value of our intellectual property or narrow the scope of our future owned and licensed patents.
In addition, the patent positions of companies in the development and commercialization of pharmaceuticals are particularly uncertain. In the United States, recent rulings from the U.S. Supreme Court and the Court of Appeals for the Federal Circuit have narrowed the scope of patent protection available in specified circumstances and weakened the rights of patent owners in specified situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts in the United States and the USPTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we or our licensors might obtain in the future. We cannot predict how future decisions by the federal courts in the United States, the U.S. Congress or the USPTO may impact the value of our patents. Any similar adverse change in the patent laws of other jurisdictions could also adversely affect our business, financial condition, results of operations and prospects.
For our U.S. patent applications, which contain claims entitled to priority date after March 16, 2013, there is a greater level of uncertainty due to the Leahy-Smith America Invents Act (the “Leahy-Smith Act”), signed into law on September 16, 2011. The Leahy-Smith Act included a number of significant changes to U.S. patent law. These included provisions that affect the way patent applications are prosecuted, redefine prior art and provide more efficient and cost-effective avenues for competitors to challenge the validity of patents. The USPTO has promulgated regulations and developed procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, did not come into effect until March 16, 2013. The Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
An important change introduced by the Leahy-Smith Act is that, as of March 16, 2013, the United States transitioned to a “first-to-file” system for deciding which party should be granted a patent when two or more patent applications are filed by different parties claiming the same invention. This requires us to be cognizant of the time from invention to filing of a patent application. Furthermore, our ability
- 46 -
to obtain and maintain valid and enforceable patents depends on whether the differences between our technology and the prior art allow our technology to be patentable over the prior art. Since patent applications in the United States and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to either: (i) file any patent application related to our product candidates or (ii) invent any of the inventions claimed in our patents or patent applications.
Among some of the other changes introduced by the Leahy-Smith Act are changes that limit where a patentee may file a patent infringement suit and new procedures providing opportunities for third parties to challenge any issued patent in the USPTO. These new post grant challenges include post grant review and inter partes review proceedings before the Patent Trial and Appeal Board at the USPTO. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in U.S. federal court necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. However, recent changes at the USPTO have resulted in many fewer inter partes review proceedings being instituted, and the USPTO has proposed modifications to the rules of practice for implementing inter partes review proceedings. The proposed modifications, if adopted, could impact the ability of third parties, including us, to challenge the validity of granted U.S. patents before the USPTO.
Geopolitical actions in the United States and in foreign countries could increase the uncertainties and costs surrounding the prosecution or maintenance of patent applications and the maintenance, enforcement or defense of issued patents. For example, the United States and foreign government actions related to Russia’s invasion of Ukraine resulted in Russia issuing Decree No. 299 that effectively nullifies the enforcement of Russian patents owned by entities and individuals in “unfriendly” countries, including the United States.
Similarly, changes in patent law and regulations in other countries or jurisdictions or changes in the governmental bodies that enforce them or changes in how the relevant governmental authority enforces patent laws or regulations may weaken our ability to obtain new patents or to enforce patents that we have licensed or that we may obtain in the future.
Any trademarks we have obtained or may obtain may be infringed or otherwise violated, or successfully challenged. If our trademarks and trade names are not adequately protected, or if we are unable to obtain desired trademarks or trade names, then we may not be able to build brand name recognition in our markets of interest and our business may be adversely affected.
We expect to rely on trademarks as one means to distinguish our product candidates, if approved for marketing, from third-party products. Once we select new trademarks and apply to register them, our trademark applications may not be approved. During trademark registration proceedings in the United States and foreign jurisdictions, we may receive rejections. We are given an opportunity to respond to those rejections, but we may not be able to overcome such rejections.
We have also not yet registered trademarks for any of our product candidates in any jurisdiction. Any trademark applications we file may be rejected and registered trademarks may not be obtained, maintained or enforced. If we do not successfully register our trademarks, we may encounter difficulty in enforcing, or be unable to enforce, our trademark rights against third parties, which could adversely affect our business and our ability to effectively compete in the marketplace.
In addition, any proprietary name we propose to use with any of our product candidates in the United States will need to be approved by the FDA, regardless of whether we have registered, or applied to register, the proposed proprietary name as a trademark. The FDA conducts a review of proposed proprietary names, including an evaluation of potential for confusion with other products’ proprietary names, as part of the biologics license application review process. If the FDA objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable proprietary name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA.
In addition, our unregistered trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on, misappropriating or violating other marks. In the USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademark registrations may not survive such proceedings. In the event that our trademarks are successfully challenged, we could be forced to rebrand our product candidates, which could result in loss of brand recognition and could require us to devote resources to advertising and marketing new brands. At times, competitors may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion.
- 47 -
Our competitors may also infringe or otherwise violate our trademarks and we may not have adequate resources to enforce our trademarks. We may not be able to protect our rights to our trademarks and trade names, which we need to build name recognition among potential collaborators or customers in our markets of interest. Any of the foregoing events may have a material adverse effect on our business.
Furthermore, in many countries, owning and maintaining a trademark registration may not provide an adequate defense against a subsequent infringement claim asserted by the owner of a senior trademark. Over the long term, if we are unable to successfully register our trademarks and trade names and establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively, and our business may be adversely affected. Our efforts to enforce or protect our proprietary rights related to trademarks, trade names, domain names or other intellectual property may be ineffective and could result in substantial costs and diversion of resources and could adversely impact our financial condition or results of operations.
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit it to maintain our competitive advantage. For example:
| ● | our product candidates, if approved, may eventually become commercially available in generic or biosimilar product forms; |
| ● | others may be able to make similar molecules to our product candidates that are not covered by the claims of the patents that we license or own now or in the future; |
| ● | we, or current or future licensors or collaborators, might not have been the first to file patent applications covering certain of our or their inventions, potentially resulting in the invalidation of such patents or refusal of such applications; |
| ● | we, or current or future licensors or collaborators, might not have been the first to make the inventions covered by the issued patent or pending patent application that we license or own now or in the future; |
| ● | we, or current or future licensors or collaborators, may fail to meet our obligations to the U.S. government regarding any patents and patent applications funded by U.S. government grants, leading to the loss or unenforceability of patent rights; |
| ● | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing on our owned or licensed intellectual property rights; |
| ● | it is possible that our pending patent applications or those that we may own or license in the future will not lead to issued patents; |
| ● | it is possible that there are prior public disclosures that could invalidate our patents; |
| ● | it is possible that there are unpublished patent applications that may later issue with claims covering our product candidates or technology similar to ours; |
| ● | it is possible that our patents or patent applications omit individual(s) that should be listed as inventor(s) or include individual(s) that should not be listed as inventor(s), which may cause these patents or patents issuing from these patent applications to be held invalid or unenforceable or result in a change in ownership; |
| ● | issued patents to which we hold rights may be held invalid, unenforceable or narrowed in scope, including as a result of legal challenges; |
| ● | the claims of our issued patents or patent applications, if and when issued, may not cover our product candidates or narrowly cover them in such a way that competitors may be able to design around to avoid infringement allegations; |
| ● | the laws of foreign countries may not protect our proprietary rights or the proprietary rights of current or future licensors or collaborators to the same extent as the laws of the United States; |
| ● | the inventors of our patents or patent applications may become involved with competitors, develop products or processes that are similar to or alternative to those claimed in our patent filings or become hostile to our patents or patent applications on which they are named as inventors; |
| ● | our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| ● | we have engaged in scientific collaborations in the past and we intend to continue to do so in the future, and our collaborators may develop adjacent or competing products that are outside the scope of our patents; |
| ● | we may not develop additional proprietary technologies that are patentable; |
| ● | the product candidates we develop may be covered by third-party patents or other intellectual property rights; |
| ● | the patents of others may prohibit or otherwise harm our ability to conduct our business; or |
| ● | we may choose not to file a patent in order to maintain certain know-how, and a third party may subsequently commercialize the technology and/or file a patent covering such intellectual property. |
- 48 -
Risks Related to Regulatory Approval of the Company’s Product Candidates and Other Legal Compliance Matters
The Company’s future product candidates and the activities associated with their development and commercialization, including their design, testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by the FDA, Health Canada and by comparable authorities in other countries.
If the Company, one of its contractors, or license partners are not able to comply with regulations and guidelines governing pharmaceutical product development (including, but not limited to GMP, Good Clinical Practices, GLP, quality assurance/quality control, and guidelines set forth by the International Conference for Harmonization), it could impact the overall development and/or commercialization activities, the timing of development or result in a supply disruption of commercial product that would negatively impact the business.
The development and manufacturing of pharmaceutical products is strictly governed by a series of standardized regulations and guidelines to ensure data and product quality including, but not limited to GMP, GLP, and additional guidelines set forth by the International Conference for Harmonization. These guidelines are mandatory standards for most regulatory agencies and designed to ensure the highest quality of research and manufacturing for pharmaceutical products. The Company’s management team has experience in the development and commercialization of pharmaceutical products under these regulations. The Company aims to put in place infrastructure to ensure compliance with relevant guidelines, including standard operation procedures and third-party audits. Despite these precautions, it is possible that activities conducted internally or by a third party may be non-compliant with industry standard regulations, with significant negative impact on the Company.
During product development, non-compliance with standard guidelines and regulations may invalidate drug product and/or data such that they are not appropriate to support regulatory filings. The Company may be required to repeat development activities as a result, incurring additional development risk and costs. Repeating specific development activities could also delay overall development and commercialization timelines, negatively impacting a product’s revenue potential. Adverse effects on timing and costs could lead to discontinuation of product development. In the event that non-compliance with standard guidelines adversely impacts clinical trial activities and trial participants, the Company could also be exposed to substantial reputational risk and legal liabilities. Regardless of merit or eventual outcome, liability claims may result in decreased demand for any product candidates or products that it may develop, injury to the Company’s reputation and significant negative media attention, significant costs to defend the related litigation, substantial monetary awards to trial participants, loss of revenue and the inability to commercialize any products that the Company may develop.
For commercial products, non-compliance with standard guidelines and regulations may prevent the Company from releasing product to the market or require the Company to withdraw product from the market. In either case, the Company would incur manufacturing costs for product without the potential to generate revenues. In addition, delays in delivery of product to the market could adversely impact long-term product utilization and drive substitution to competitor products. In the case where product released to the market is retroactively found to be non-compliant with existing guidelines, the Company could also incur significant costs related to the returns, refunds, and destructions of non-compliant product. Additionally, the Company could be exposed to substantial reputational risk and legal liabilities with potential negative consequences outlined above.
In any situation of guideline non-compliance, the Company will be required to undertake a comprehensive investigation and engage in activities to remedy and prevent future deviations. These activities could impose significant costs on the Company and draw resources away from other Company objectives.
Failure to obtain regulatory approval in international jurisdictions would prevent the Company’s product candidates from being marketed abroad. Risk of a rejection, an incomplete response, a poor approved label or pricing restrictions by a regulatory authority outside of the United States may adversely impact the United States market opportunity and limit the value of the asset to the Company.
The Company intends to enter into agreements with third parties for the marketing of its products outside Canada and the United States. In order to market and sell the Company’s products in the European Union and many other jurisdictions, the Company or its third parties must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA or Health Canada approval. The regulatory approval process outside the United States generally includes all the risks associated with obtaining FDA or Health Canada approval. In addition, in many countries outside the United States or Canada, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. The Company
- 49 -
may not obtain approvals from regulatory authorities outside the United States or Canada on a timely basis, if at all. Approval by the FDA or Health Canada does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the United States or Canada does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. The Company may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize its products in any market, thus limiting its revenue potential.
The Company’s direct and indirect relationships with healthcare customers, government, payors, and reimbursement/contract decision makers, will be subject to applicable anti-bribery, anti-corruption and other healthcare laws and regulations, which could expose the Company to criminal sanctions, civil penalties, program exclusion, debarment, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescription of any product candidates for which the Company obtains marketing approval. The Company’s future arrangements with third-party payors and customers may expose the Company to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which it markets, sells and distributes its products for which it obtains marketing approval. Restrictions under applicable United States federal and state healthcare laws and regulations that may impact the Company’s activities, include the following:
| ● | the federal healthcare anti-kickback statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs; |
| ● | civil penalties could be imposed against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
| ● | criminal and civil liability could be imposed for executing a scheme to defraud any healthcare benefit program and also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| ● | manufacturers of drugs, devices, biologics and medical supplies are generally required to report information related to physician payments and other transfers of value and physician ownership and investment interests; and |
| ● | analogous laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, and some laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures. |
Costs will be substantial to ensure that the Company’s business arrangements with third parties will comply with applicable healthcare laws and regulations in each jurisdiction where the Company products will eventually be offered. It is possible that governmental authorities will conclude that the Company’s business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If the Company’s operations are found to be in violation of any of these laws or any other governmental regulations that may apply to it, it may be subject to significant civil, criminal and administrative penalties, damages, fines, exclusion from government funded healthcare programs, such as Medicare and Medicaid in the United States, and the curtailment or restructuring of the Company’s operations. If any of the physicians or other providers or entities with whom the Company expects to do business are found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Market access, legislative and pricing policy changes may increase the difficulty and cost for the Company to obtain optimal marketing approval to commercialize its product candidates and affect the prices it may obtain.
In the United States and other foreign jurisdictions, there have been legislative and regulatory changes and proposed changes regarding, among other matters, biopharmaceutical product approvals and pricing that could prevent or delay marketing approval of and commercial prospects for the Company’s product candidates, potentially having a negative impact on its ability to profitably sell any of its future product candidates. Such an outcome may have a materially adverse effect on the value and going concern prospects of the Company.
- 50 -
Other Risks
Anticipated changes to economic or geo-political conditions may impact the Company.
The Company, its operations, its plans and its ability to raise financing may be adversely affected in subsequent fiscal periods as a result of current and future geopolitical events, including as a result of risks and uncertainties surrounding potential regulatory changes or the establishment of protectionist measures, such as the imposition of tariffs or modifications to free trade agreements.
The policies adopted by President Trump may result in legislative and regulatory changes that could have an adverse effect on the Company and its financial condition. In particular, there is uncertainty regarding U.S. tariffs and support for existing treaty and trade relationships, including with Canada. Implementation by the U.S. government of new legislative or regulatory policies could impose additional production costs on the Company, decrease U.S. demand for the Company’s products, or otherwise negatively impact the Company, which may have a material adverse effect on the Company’s business, financial condition, and operations. In addition, this uncertainty may adversely impact: (i) the ability of U.S. companies to transact business with Canadian companies; (ii) the Company’s profitability; (iii) regulation affecting the Company’s industry; (iv) global stock markets (including the TSX); and (v) general global economic conditions. All of these factors are outside of the Company’s control but may nonetheless lead the Company to adjust its strategy in order to compete effectively in global markets.
We are subject to intense competition for skilled personnel. The loss of key personnel or the inability to attract additional personnel could impair our ability to conduct operations.
We are highly dependent on our management and staff; the loss of whose services might adversely impact our ability to achieve our objectives. Recruiting and retaining qualified management and other personnel is critical to our success. Competition for skilled personnel is intense, and the ability to attract and retain qualified personnel may be affected by such competition. We do not maintain “key person” insurance for any of our key personnel. The Company also depends on scientific and clinical collaborators and advisors, all of whom have outside commitments that may limit their availability. The Company has expanded significantly over the past twelve months and will continue to add personnel should SAT-3247 advance through the various stages of clinical development. This expansion may result in difficulties managing growth, which could disrupt the Company’s operations.
This growth will also result in significant additional responsibilities on management, who may need to spend a disproportionate amount of its attention away from the business operations and spend a substantial amount of time to managing these growth activities. Managing this growth will also require the Company to continue to implement and improve its managerial, operational, and financial systems, and continue to recruit and train additional qualified personnel. Due to its limited financial resources, the Company may not be able to effectively manage the expansion of its operations or recruit and train additional qualified personnel. If the Company is unable to effectively manage this growth, the expenses may increase more than anticipated and the Company may not be able to effectively implement its business strategy.
Failure to maintain adequate internal controls over financial processes and reporting may negatively impact the Company’s results of operations or its ability to comply with its reporting obligations.
Effective internal controls are necessary for the Company to provide reliable financial reports and to help prevent fraud. Although the Company intends to undertake a number of procedures in order to help ensure the reliability of its financial reports, including those imposed on it under Canadian securities laws, the Company cannot be certain that such measures will ensure that the Company will maintain adequate control over financial processes and reporting. Failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm the Company’s results of operations or cause it to fail to meet its reporting obligations.
As a Nasdaq-listed reporting issuer in the United States, we are subject to certain of the requirements of the Sarbanes-Oxley Act of 2002, as amended (“SOX”). Section 404 of SOX (“Section 404”) requires companies subject to the reporting requirements of the U.S. securities laws to complete a comprehensive evaluation of its internal controls over financial reporting. To comply with this statute, we are required to document and test our internal control procedures and our management is required to assess and issue a report concerning the Company’s internal controls over financial reporting. Pursuant to the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”), we are classified as an “emerging growth company” and are exempt from certain reporting requirements, including the independent auditor attestation requirements of Section 404(b) of SOX. Under this exemption, our independent auditor is not required to attest to and report on management’s assessment of our internal controls over financial reporting so long as we remain an emerging growth company. We need to prepare for compliance with Section 404 by strengthening, assessing and testing our system of internal controls
- 51 -
to provide the basis for the report. However, the continuous process of strengthening our internal controls and complying with Section 404 is complicated and time-consuming. Furthermore, we believe that our business will grow both domestically and internationally, in which case our internal controls will become more complex and will require significantly more resources and attention to ensure our internal controls remain effective overall. During the course of our testing, management may identify material weaknesses or significant deficiencies, which may not be remedied in a timely manner to meet the deadline imposed by SOX. If management cannot favorably assess the effectiveness of our internal controls over financial reporting, or our independent registered public accounting firm identifies material weaknesses in our internal controls, investor confidence in our financial results may weaken, and the market price of our securities may suffer.
Our operations could be adversely affected by events outside of our control, such as natural disasters, wars or health epidemics.
We may be impacted by business interruptions resulting from pandemics and public health emergencies, geopolitical actions, including war and terrorism or natural disasters including earthquakes, typhoons, floods, and fires. An outbreak of infectious disease, a pandemic or a similar public health threat, or a fear of any of the foregoing, could adversely impact us by causing operating, manufacturing supply chain, clinical trial and project development delays and disruptions, labor shortages, travel, and shipping disruption and shutdowns (including as a result of government regulation and prevention measures). It is unknown whether and how we may be affected if such an epidemic persists for an extended period of time. We may incur expenses or delays relating to such events outside of our control, which could have a material adverse impact on our business, operating results, and financial condition.
Our Director and Officer (“D&O”) insurance policies may be inadequate and potentially expose us to unrecoverable risks.
We have limited D&O and commercial insurance policies. Any significant insurance claims could have a material adverse effect on our financial condition. Insurance availability, coverage terms and pricing vary with market conditions. We will continue to obtain appropriate insurance coverage for insurable risks that we identify; however, we may fail to correctly anticipate or quantify these risks, or we may not be able to obtain appropriate insurance coverage, or insurers may not cover insurable events that may occur as we expect. Conditions in the insurance markets are rapidly changing including those related to director and officer coverage. These conditions have resulted in rising premium costs, higher policy deductibles and lower coverage limits. For some risks, we may not be able to maintain insurance coverage because of cost or availability.
We may incur losses associated with foreign currency fluctuations.
Our functional and reporting currency is the Canadian dollar and some of our operations are conducted in currencies other than the Canadian dollar (principally in U.S. dollars) and a portion of our net monetary assets are denominated in other currencies (principally in U.S. dollars). Inflation in the United States may have the effect of increasing the Canadian dollar cost of our operations. If the Canadian dollar declines in value in relation to the U.S. dollar or other currencies, it will become more expensive for us to fund our operations. Similarly, fluctuations in the value of foreign currencies relative to the Canadian dollar could cause us to incur currency exchange losses.
Our information technology (“IT”) and infrastructure may fail or suffer security breaches, which could result in significant disruption of our discovery and product development programs and our ability to operate our business effectively.
Satellos contracts IT services through internet service, web hosting and data sharing service providers. We also engage with a wide range of collaborators, contractors or consultants, financial institutions and other business providers or partners. It is possible that any of our systems, whether in-house or contracted, and those of our current and future collaborators, contractors or consultants, financial institutions and other business providers or partners may be vulnerable to disruptive elements, including computer viruses, phishing attacks, espionage and hacking programs resulting in unauthorized access; or be damaged or compromised through natural disasters, terrorism, war and telecommunication or electrical failures. Although we take such steps to help protect sensitive information from unauthorized access or disclosure, our IT and infrastructure may be vulnerable in the future to attacks by hackers or viruses, failures, or breaches due to third-party action, employee or contractor negligence or error, malfeasance, or other incidents or disruptions. If such an event were to occur and cause interruptions in our operations or result in a loss of or damage to, our data, or inappropriate access to or disclosure of confidential or proprietary information including trade secrets, Satellos could incur liability, our competitive position could be harmed, our reputation could be damaged, and the further development and commercialization of our product candidates could be significantly compromised or delayed.
- 52 -
Risks Related to our Securities
The price of our Common Shares has experienced volatility and may be subject to fluctuation in the future based on market conditions.
The market prices for the securities of biotechnology companies, including our own, have historically been highly volatile. The market has from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of any particular company. In addition, because of the nature of our business, certain factors such as our announcements, competition from new therapeutic products or technological innovations, government regulations, fluctuations in our operating results, results of clinical trials, public concern regarding the safety of drugs generally, general market conditions and developments in patent and proprietary rights can have an adverse impact on the market price of our Common Shares. For example, from January 1, 2025 through December 31, 2025, the price of the Common Shares on the TSX ranged from a low of Cdn$6.24 to a high of Cdn$11.28. Any change in the public’s perception of our prospects could cause the price of our listed securities to change dramatically. Furthermore, any negative change in the public’s perception of the prospects of biotechnology companies in general or the market in general could depress our share price regardless of our results. Volatility or depression in the capital markets, particularly with respect to biotechnology stocks, could also affect our ability to raise additional capital.
An active trading market for the Common Shares may not be sustained.
Although Satellos has listed its Common Shares on the TSX and Nasdaq, an active trading market for the Common Shares may not be sustained. If an active trading market for the Common Shares is not maintained, the liquidity of the Common Shares and the prices that may be obtained for the Common Shares will be adversely affected. The average number of shares traded in any given day over the past year has been relatively small compared to the public float. Thus, the actions of a few shareholders either buying or selling the Common Shares may adversely affect the price of the Common Shares. Historically, securities similar to the Common Shares have experienced extreme price and volume fluctuations that do not necessarily relate to operating performance and could result in rapid and substantial losses for shareholders.
Our shareholders may experience significant dilution from future sales of our securities.
We anticipate that we will need to raise additional capital in the future. The sale of additional equity, including Pre-Funded Warrants, warrants or debt securities, if convertible into equity, and including the 2026 Offering, will result in dilution to our existing shareholders. Also, any debt financing, if available, may require us to pledge our assets as collateral or involve restrictive covenants, such as limitations on our ability to incur additional indebtedness, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could negatively impact our ability to conduct our business. As a result, our net income per share could decrease in future periods and the market price of our Common Shares could decline. The perceived risk of dilution may negatively impact the price of our shares and may cause our shareholders to sell their shares, which would contribute to a decline in the price of the Common Shares. Moreover, the perceived risk of dilution and the resulting downward pressure on our share price could encourage investors to engage in short sales of the Common Shares, which could further contribute to progressive price declines in the Common Shares.
Satellos may be subject to securities litigation, which is expensive and could divert management attention.
The market price of the Common Shares may be volatile, and in the past companies that have experienced volatility in the market price of their shares have been subject to securities class action litigation. Satellos may be the target of this type of litigation in the future. Litigation of this type could result in substantial costs and diversion of management’s attention and resources, which could adversely impact Satellos’ business. Any adverse determination in litigation could also subject Satellos to significant liabilities.
Satellos has never paid dividends on the Common Shares and Satellos does not anticipate paying any dividends in the foreseeable future. Consequently, any gains from an investment in the Common Shares will likely depend on whether the price of the Common Shares increases.
Satellos has not paid dividends on the Common Shares to date and Satellos currently intends to retain Satellos’ future earnings, if any, to fund the development and growth of Satellos’ business. As a result, capital appreciation, if any, of the Common Shares will be your sole source of gain for the foreseeable future. Consequently, in the foreseeable future, you will likely only experience a gain from your investment in the Common Shares if the price of the Common Shares increases.
- 53 -
If equity research analysts do not publish research or reports about Satellos’ business or if they issue unfavourable commentary or downgrade the Common Shares, the price of the Common Shares could decline.
The trading market for the Common Shares will rely in part on the research and reports that equity research analysts publish about Satellos and Satellos’ business. Satellos does not control these analysts. The price of the Common Shares could decline if one or more equity analysts downgrade the Common Shares or if analysts issue other unfavourable commentary or cease publishing reports about Satellos or Satellos’ business.
We may be or become a passive foreign investment company, which could result in adverse U.S. federal income tax consequences to U.S. Holders.
The rules governing passive foreign investment companies (“PFICs”) can have adverse effects for U.S. federal income tax purposes. The tests for determining PFIC status for a taxable year depend upon the relative values of certain categories of assets and the relative amounts of certain kinds of income. The determination of whether we are a PFIC, which must be made annually after the close of each taxable year, depends on the particular facts and circumstances (such as the valuation of our assets, including goodwill and other intangible assets) and may also be affected by the application of the PFIC rules, which are subject to differing interpretations. Moreover, our ability to earn specific types of income that we currently treat as non-passive for purposes of the PFIC rules is uncertain with respect to future years. Based on our gross income, the average value of our assets, including goodwill and the nature of our active business, it is possible that we would be treated as a PFIC for U.S. federal income tax purposes for the taxable year ending December 31, 2025, although a complete PFIC analysis has not been performed. Furthermore, there can be no assurance that we will not be considered a PFIC for our current taxable year ending December 31, 2026, or for any future taxable year.
If we are a PFIC, a U.S. shareholder would be subject to adverse U.S. federal income tax consequences, such as ineligibility for any preferred tax rates on capital gains or on actual or deemed dividends, interest charges on certain taxes treated as deferred, and additional reporting requirements under U.S. federal income tax laws and regulations. A U.S. shareholder may in certain circumstances mitigate adverse tax consequences of the PFIC rules by filing an election to treat the PFIC as a qualified electing fund (“QEF”) or, if shares of the PFIC are “marketable stock” for purposes of the PFIC rules, by making a mark-to-market election with respect to the shares of the PFIC. A U.S. shareholder who makes a QEF election generally must report on a current basis its share of the Company’s net capital gain and ordinary earnings for any year in which the Company is a PFIC, whether or not the Company distributes any amounts to its shareholders. However, U.S. shareholders should be aware that in the event that we are a PFIC and a U.S. shareholder wishes to make a QEF election, we can make no assurances that we will be able to supply U.S. shareholders with information that such U.S. shareholders require to make a QEF election. If a U.S. shareholder makes a mark-to-market election with respect to its shares, the U.S. shareholder is required to include annually in its U.S. federal taxable income an amount reflecting any year end increase in the value of its shares. Investors should consult their own tax advisors regarding all aspects of the application of the PFIC rules to the shares.
U.S. investors may not be able to obtain enforcement of civil liabilities against us.
The enforcement by investors of civil liabilities under the U.S. federal or state securities laws may be affected adversely by the fact that we are governed by the CBCA, that the majority of our officers and directors are residents of Canada or otherwise reside outside the United States, and that all, or a substantial portion of their assets and a substantial portion of our assets, are located outside the United States. It may not be possible for investors to effect service of process within the United States on certain of our directors and officers or enforce judgments obtained in the United States courts against us or certain of our directors and officers based upon the civil liability provisions of United States federal securities laws or the securities laws of any state of the United States.
There is some doubt as to whether a judgment of a U.S. court based solely upon the civil liability provisions of U.S. federal or state securities laws would be enforceable in Canada against us or our directors and officers. There is also doubt as to whether an original action could be brought in Canada against us or our directors and officers to enforce liabilities based solely upon U.S. federal or state securities laws.
As a foreign private issuer, we are subject to different U.S. securities laws and rules than a domestic U.S. issuer, which may limit the information publicly available to our U.S. shareholders
We are a foreign private issuer under applicable U.S. federal securities laws and, therefore, we are not required to comply with all the periodic disclosure and current reporting requirements of the U.S. Exchange Act, and related rules and regulations, applicable to U.S. domestic issuers. We do not file all of the same reports that a U.S. domestic issuer would file with the SEC, although we are required to
- 54 -
file with or furnish to the SEC the continuous disclosure documents that we are required to file in Canada under Canadian securities laws. In addition, as a foreign private issuer, we are exempt from the proxy rules under the U.S. Exchange Act.
As a foreign private issuer, we are exempt from the rules and regulations under the U.S. Exchange Act related to the furnishing and content of proxy statements. We are also exempt from Regulation FD, which prohibits issuers from making selective disclosures of material non-public information. While we expect to comply with the corresponding requirements relating to proxy statements and disclosure of material non-public information under Canadian securities laws, these requirements may differ from those under the U.S. Exchange Act and Regulation FD and shareholders should not expect to receive in every case the same information at the same time as such information is provided by U.S. domestic companies.
In addition, as a foreign private issuer, we have the option to follow certain Canadian corporate governance practices, except to the extent that such laws would be contrary to U.S. securities laws, and provided that we disclose the requirements we are not following and describe the Canadian practices we follow instead. For example, we do not intend to follow the minimum quorum requirements for shareholder meetings, as well as certain Nasdaq shareholder approval requirements prior to the issuance of securities, as permitted for foreign private issuers. As a result, our shareholders may not have the same protections afforded to shareholders of U.S. domestic companies that are subject to all U.S. corporate governance requirements.
Following the completion of this offering, we may cease to qualify as a foreign private issuer. If we cease to qualify, we will be subject to the same reporting requirements and corporate governance requirements as a U.S. domestic issuer which may increase our costs of being a public company in the United States.
We may lose our foreign private issuer status in the future, which could result in significant additional costs and expenses to us.
In order to maintain our current status as a foreign private issuer, a majority of our Common Shares must be either directly or indirectly owned by non-residents of the United States unless we also satisfy one of the additional requirements necessary to preserve this status. We may in the future lose our foreign private issuer status if a majority of the Common Shares are held in the United States and we fail to meet the additional requirements necessary to avoid loss of foreign private issuer status. The regulatory and compliance costs to us under U.S. federal securities laws as a U.S. domestic issuer may be significantly more than the costs we incur as a Canadian foreign private issuer eligible to use the multijurisdictional disclosure system (“MJDS”). If we are not a foreign private issuer, we would not be eligible to use the MJDS or other foreign issuer forms and would be required to file periodic and current reports and registration statements on U.S. domestic issuer forms with the SEC, which are more detailed and extensive than the forms available to a foreign private issuer. We may lose the ability to rely upon exemptions from Nasdaq corporate governance requirements that are available to foreign private issuers.
If a U.S. holder is treated as owning at least 10% of our Common Shares, such U.S. holder may be subject to adverse U.S. federal income tax consequences.
If a U.S. holder is treated as owning, directly, indirectly or constructively, at least 10% of the value or voting power of our Common Shares, such U.S. holder may be treated as a “United States shareholder” with respect to each “controlled foreign corporation” in our group, if any. Generally, a non-U.S. corporation is deemed as a controlled foreign corporation if more than 50% of its stock (by voting power or value) is owned (directly, indirectly or constructively) by U.S. shareholders. We will generally be classified as a controlled foreign corporation if more than 50% of our outstanding shares, measured by reference to voting power or value, are owned (directly, indirectly or by attribution) by U.S. shareholders. In addition, since our group includes one or more U.S. subsidiaries, certain of our non-U.S. subsidiaries could be treated as controlled foreign corporations, regardless of whether we are treated as a controlled foreign corporation. If we or any of our non-U.S. subsidiaries are treated as controlled foreign corporations, a U.S. shareholder would be required to annually report and include in its U.S. taxable income its pro rata share of “Subpart F income,” “global intangible low-taxed income” and investments in U.S. property by the controlled foreign corporation, regardless of whether we make any distributions, and generally would not be allowed certain tax deductions or foreign tax credits that would be allowed to a U.S. shareholder of a U.S. corporation. In addition, failure to comply with certain reporting obligations may subject a U.S. shareholder to monetary penalties and may prevent the statute of limitations with respect to such shareholder’s U.S. federal income tax return for the year for which reporting was due from starting. We cannot provide any assurances that we will assist our investors in determining whether any of our non-U.S. subsidiaries are treated as a controlled foreign corporation or whether such investor is treated as a U.S. shareholder with respect to any of such controlled foreign corporations. Further, we cannot provide any assurances that we will furnish to any U.S. shareholder information that may be necessary to comply with the reporting and tax paying obligations described in this risk factor. U.S. holders should consult their tax advisors regarding the potential application of these rules to their investment in our Common Shares.
- 55 -
We are an emerging growth company and intend to rely on and take advantage of reduced disclosure requirements applicable to emerging growth companies, which could make our Common Shares less attractive to investors
We are an “emerging growth company” as defined in the JOBS Act and Section 3(a) of the U.S. Exchange Act, and we will continue to qualify as an emerging growth company until the earliest to occur of: (a) the last day of the fiscal year during which we have total annual gross revenues of US$1,235,000,000 (as such amount is indexed for inflation every five years by the SEC) or more; (b) the last day of the fiscal year of the Company following the fifth anniversary of the date of the first sale of common equity securities of our company pursuant to an effective registration statement under the U.S. Securities Act; (c) the date on which we have, during the previous three year period, issued more than US$1,000,000,000 in non-convertible debt; and (d) the date on which we are deemed to be a “large accelerated filer”, as defined in Rule 12b-2 under the U.S. Exchange Act. We will qualify as a “large accelerated filer” under the rules of the SEC (and would cease to be an emerging growth company) at such time when on the last business day of its second fiscal quarter of such year the aggregate worldwide market value of our common equity held by non-affiliates will be US$700,000,000 or more. For so long as we remain an emerging growth company, we are permitted to and intend to rely upon exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act (2002), as amended. We cannot predict whether investors will find the Common Shares less attractive because we rely upon certain of these exemptions. If some investors find the Common Shares less attractive as a result, there may be a less active trading market for the Common Shares and the Common Share price may be more volatile. On the other hand, if we no longer qualify as an emerging growth company, we would be required to divert additional management time and attention from our development and other business activities and incur increased legal and financial costs to comply with the additional associated reporting requirements, which could negatively impact our business, financial condition and results of operations.
- 56 -
The Company has not, since its inception, declared or paid any dividends on its Common Shares. The declaration of dividends on our Common Shares is within the discretion of the Board and will depend on the assessment of, among other factors, capital requirements, earnings, and the operating and financial condition of the Company. At the present time, the Company’s anticipated capital requirements are such that Satellos follows a policy of retaining all available funds and any future earnings in order to finance its technology advancement, business development and corporate growth. Satellos does not intend to declare or pay cash dividends on its Common Shares within the foreseeable future. See “Risk Factors – Risks Related to Our Securities – Satellos has never paid dividends on its Common Shares and Satellos does not anticipate paying any dividends in the foreseeable future”.
- 57 -
DESCRIPTION OF CAPITAL STRUCTURE
Common Shares
The authorized share capital of the Company is an unlimited number of Common Shares without par value. As at December 31, 2025, the Company had 15,458,903 Common Shares issued and outstanding as fully paid and non- assessable. All of our Common Shares are of the same class and, once issued, rank equally. The holders of Common Shares are entitled to dividends, if, as and when declared by the Board, to one vote per Common Share at meetings of the shareholders of the Company and, upon liquidation, to share equally in such assets of the Company as are distributable to the holders of Common Shares. There are no pre-emptive or conversion rights.
Pre-Funded Warrants
As at December 31, 2025, the Company had 3,159,743 Pre-Funded Warrants issued and outstanding, exercisable into an equal number of Common Shares of Satellos for Cdn$0.00012 per Pre-Funded Warrant with no expiry date.
Stock Options
The Company has a stock option plan which authorizes Satellos to grant options to acquire Common Shares (“Options”) to directors, officers, employees and consultants of Satellos or any of its subsidiaries. The Company’s option plan is an evergreen option plan whereby the Company is authorized to grant Options under the plan, not to exceed 15% of the Company’s total issued and outstanding shares. As of December 31, 2025, there were 2,202,960 Options outstanding under the Company’s option plan. For more details, see the Company’s management information circular dated May 12, 2025, for its annual general and special meeting of Shareholders held on June 18, 2025, which attaches the Company’s option plan.
- 58 -
Trading Price and Volume
During the year ended December 31, 2025, the Common Shares of the Company were listed for trading on the TSX under the symbol “MSCL”. The following table sets forth the high and low trading prices and the volume of shares traded on the TSX for the Common Shares for each month in 2025.
Month | | High | | Low | | Volume |
January |
| Cdn$10.68 |
| Cdn$9.36 |
| 138,104 |
February |
| Cdn$10.56 |
| Cdn$8.64 |
| 420,844 |
March |
| Cdn$11.28 |
| Cdn$8.64 |
| 291,837 |
April |
| Cdn$9.00 |
| Cdn$6.84 |
| 252,670 |
May |
| Cdn$9.12 |
| Cdn$7.32 |
| 301,142 |
June |
| Cdn$8.28 |
| Cdn$6.24 |
| 317,051 |
July |
| Cdn$8.40 |
| Cdn$6.36 |
| 181,172 |
August |
| Cdn$7.56 |
| Cdn$6.36 |
| 204,062 |
September |
| Cdn$8.88 |
| Cdn$7.20 |
| 831,134 |
October |
| Cdn$10.32 |
| Cdn$8.04 |
| 289,907 |
November |
| Cdn$9.72 |
| Cdn$8.04 |
| 220,684 |
December |
| Cdn$8.64 |
| Cdn$7.56 |
| 297,645 |
Prior Sales
The following table summarizes issuances of Common Shares and securities convertible or exchangeable into Common Shares during the financial year ended December 31, 2025.
| | | | Issuance/Exercise Price | | Number of Securities |
Date of Issuance | | Type of Security | | Per Security | | Issued |
January 8, 2025 |
| Warrants Exercised(2) |
| Cdn$7.20 |
| 416 |
January 9, 2025 |
| Stock Options Issued(1) |
| Cdn$10.08 |
| 81,548 |
February 10, 2025 |
| Stock Options Issued(1) |
| Cdn$9.48 |
| 711,328 |
April 30, 2025 |
| Warrants Exercised(3) |
| Cdn$6.00 |
| 1,715 |
May 2, 2025 |
| Warrants Exercised(3) |
| Cdn$6.00 |
| 3,333 |
May 13, 2025 |
| Warrants Exercised(3) |
| Cdn$6.00 |
| 39,799 |
May 13, 2025 |
| Pre-Funded Warrants Exercised |
| Cdn$0.00 |
| 549,991 |
May 14, 2025 |
| Warrants Exercised(3) |
| Cdn$6.00 |
| 4,850 |
May 15, 2025 |
| Warrants Exercised(2) |
| Cdn$7.20 |
| 3,125 |
May 16, 2025 |
| Warrants Exercised(3) |
| Cdn$6.00 |
| 3,891 |
May 20, 2025 |
| Warrants Exercised(3) |
| Cdn$6.00 |
| 266,666 |
June 18, 2025 |
| Stock Options Issued(1) |
| Cdn$6.60 |
| 73,757 |
August 15, 2025 |
| Stock Options Issued(1) |
| Cdn$6.96 |
| 224,091 |
August 25, 2025 |
| Stock Options Issued(1) |
| Cdn$7.08 |
| 12,500 |
September 3, 2025 |
| Warrants Exercised(2) |
| Cdn$7.20 |
| 4,166 |
September 5, 2025 |
| Warrants Exercised(2) |
| Cdn$7.20 |
| 11,666 |
September 9, 2025 |
| Warrants Exercised(2) |
| Cdn$7.20 |
| 16,666 |
September 10, 2025 |
| Warrants Exercised(2) |
| Cdn$7.20 |
| 18,229 |
September 11, 2025 |
| Warrants Exercised(2) |
| Cdn$7.20 |
| 25,520 |
September 12, 2025 |
| Warrants Exercised(2) |
| Cdn$7.20 |
| 19,375 |
September 15, 2025 |
| Warrants Exercised(2) |
| Cdn$7.20 |
| 49,166 |
September 16, 2025 |
| Pre-Funded Warrants Exercised |
| Cdn$0.00 |
| 587,560 |
October 8, 2025 |
| Stock Options Exercised |
| Cdn$6.00 |
| 34,463 |
November 14, 2025 |
| Stock Options Issued(1) |
| Cdn$9.36 |
| 57,026 |
Notes:
(1)Granted under the stock option plan of the Company.
(2)Exercise of warrants issued on September 13, 2022 as part of a public offering.
(3)Exercise of broker warrants issued on May 17, 2023 as part of a public offering.
- 59 -
As of the date of this AIF, to the knowledge of the Company, the Company has no escrowed securities or securities subject to contractual restriction on transfer.
- 60 -
The names of the directors and executive officers of the Company as of the date hereof, their province or state and country of residence, their respective positions with our Company and the date upon which they were first elected as a director or officer of the Company are set out in the table below. The term of each director expires on the date of our next annual meeting.
| | | | | | | | No. of Common |
|
| | | | | | | | Shares Beneficially |
|
| | | | Director or Executive | | | | Owned or |
|
Name and Residence | | Position | | Officer Since | | Principal Occupation | | Controlled(4) |
|
Geoff MacKay(2) |
| Board Chair |
| July 2018 |
| CEO, Fable Therapeutics, since 2023; CEO, Avrobio, 2015 to 2023 |
| 21,201 | |
Maine, USA | | | | | | | | | |
Frank Gleeson |
| Director, President and Chief Executive Officer (“CEO”) |
| Director since July 2012, CEO since March 2018, and President since April 2018 |
| CEO, Satellos Bioscience Inc., since 2018 |
| 331,365 | |
Ontario, Canada | | | | | | | | | |
Selwyn Ho(1)(3) |
| Director | | June 18, 2025 |
| CEO, Signadori Bio, since 2025; CEO, Medigene AG, 2022 to 2025 |
| Nil | |
Berkshire, UK | | | | | | | | | |
Iris Loew-Friedrich(3) |
| Director | | June 18, 2025 |
| Self-Employed, since 2024; Chief Medical Officer, UCB SA, 2008 to 2024 |
| Nil | |
Ratingen, Germany | | | | | | | | | |
Brian Bloom |
| Director | | March 2018 |
| CEO, Bloom Burton & Co., since 2008 |
| 1,133,400 | (5) |
Ontario, Canada | | | | | | | | | |
Mark Nawacki |
| Director | | November 14, 2025 |
| President, Searchlight Pharma Inc., since 2015 |
| Nil | |
Quebec, Canada | | | | | | | | | |
Adam Mostafa(1)(2) |
| Director | | December 5, 2021 |
| CFO, Sitryx Pharmaceuticals, since 2025; CFO X4 Pharmaceuticals, 2018 to 2025 |
| Nil | |
Massachusetts, USA | | | | | | | | | |
Franklin Berger(3) |
| Director | | June 29, 2023 |
| Entrepreneur, since 2012 |
| 656,583 | |
New York, USA | | | | | | | | | |
Stephanie Brown(1) (2) |
| Director | | November 12, 2024 |
| President North America, Santhera Pharmaceutical, 2021 to 2024 |
| Nil | |
Massachusetts, USA | | | | | | | | | |
Dr. Michael Rudnicki |
| Chief Discovery Officer | | August 13, 2021 |
| Senior Scientist, OHRI, since 2000 |
| 258,159 | |
Ontario, Canada | | | | | | | | | |
Elizabeth Williams |
| Chief Financial Officer | | September 1, 2023 |
| CFO, Satellos Bioscience Inc., since 2023; CFO, Medicenna Therapeutic, 2016 to 2023 |
| 3,333 | |
Ontario, Canada | | | | | | | | | |
Dr. Phil Lambert |
| Chief Scientific Officer | | September 28, 2022 |
| CTO, Satellos Bioscience Inc., since 2022 |
| Nil | |
Maryland, USA | | | | | | | | | |
Dr. Wildon Farwell |
| Chief Medical Officer | | July 16, 2025 |
| CMO, Satellos Bioscience Inc., since 2025; CMO, Dyne Therapeutics, 2021 to 2025 |
| Nil | |
Massachusetts, USA | | | | | | | | | |
Notes:
| (1) | Member of the Audit Committee. |
| (2) | Member of the Compensation Committee. |
| (3) | Member of the Nominating and Corporate Governance Committee. |
| (4) | The information as to principal occupation and shares beneficially owned or over which control or direction is exercised is not within the knowledge of Satellos, and therefore has been furnished by each director individually. |
| (5) | 501,833 of these Common Shares are held through Bloom Burton Development Corp, an affiliated company of Bloom Burton and Co. Inc., of which Brian Bloom is co-founder, Chair and CEO, 508,334 of these Common Shares are held through Bloom Burton & Co. Inc., of which Brian Bloom is co-founder, Chair and CEO and 56,000 are held through 2194655 Ontario Inc. |
- 61 -
Share Ownership by Directors and Executive Officers
As at the date of this Annual Information Form, the current directors and officers of Satellos, as a group, beneficially owned, or controlled or directed, directly or indirectly, an aggregate of 2,404,041 Common Shares, representing approximately 15.6% of the Company’s issued and outstanding Common Shares. The information as to the number of Common Shares beneficially owned, or controlled or directed, not being within the knowledge of the Company, has been furnished by the respective directors and officers of the Company individually.
Biographical Information and Principal Occupations
The following are short biographies of the Company’s directors and executive officers:
Frank Gleeson, Director, President and Chief Executive Officer
Mr. Gleeson has been a venture capitalist, executive and entrepreneur in the biotechnology field for over 30 years. During this time, he has been a key party to building more than 20 biomedical companies from breakthrough research or technologies. Mr. Gleeson has negotiated or facilitated numerous financing, funding and M&A transactions valued in excess of $750 million. In 2018, he co-founded Satellos with Dr. Rudnicki where as CEO, through the end of Q1, 2026, he has raised $200 million, taken the company public on the TSX (MSCL) and Nasdaq (MSLE), built a team of 25 professionals, and directed the company into Phase 2 clinical development in Duchenne muscular dystrophy with its novel lead drug candidate, SAT-3247. Prior to Satellos, he and Dr. Rudnicki co-founded Verio Therapeutics, where Mr. Gleeson was CEO and managed its acquisition by Fate Therapeutics (Nasdaq: FATE). Mr. Gleeson has also served as Chief of Commercial Operations at Centre for Probe Development and Commercialization (CPDC), where he played a principal role in building a global radiopharmaceutical manufacturing business and supported the creation of two spin-out companies. Prior to CPDC, he served as an Executive-in-Residence with the Fight Against Cancer Innovation Trust (FACIT), an innovative nucleator, where he supported or led the creation, financing and monetization of three new entities. From 2000 - 2003, Mr. Gleeson was founding CEO of MDS Proteomics Inc. (MDSP), where he made and integrated three global acquisitions, built leading-edge sequencing infrastructure, a 200-person team, and raised in excess of $100 million. Prior to MDSP, he was a Senior Vice President and Venture Partner with MDS Capital Corp. (now Lumira), where he was lead partner on a fund with more than $250 million under management focused on creating drug discovery companies based on novel Canadian science. Before his tenure with MDS commencing in 1994, Mr. Gleeson enjoyed a 17-year career in operating roles of increasing scope and complexity with ICI plc (now AstraZeneca), a global chemicals, pharmaceuticals, and advanced materials company, during which he was involved in technology commercialization in several fields both in Canada and internationally. Mr. Gleeson has served on numerous boards of private and public companies and not-for-profit entities. He holds BBA (honours) and MBA degrees from York University in Toronto.
Geoff Mackay, Board Chair
Mr. MacKay has served as CEO of several innovative biotech companies over the last 20 years. He is currently CEO of a cardio-metabolic focused, pre-clinical stage company called Fable Therapeutics. Previously, he was founding CEO of AVROBIO Inc., which sold its lead asset to Novartis. Mr. MacKay was also the founding CEO of eGenesis Inc., a biotech dedicated to applying gene editing to xenotransplantation. During his tenure as CEO of Organogenesis Inc., the company received the first approval of an allogeneic cell therapy from the FDA’s Center for Biologics Evaluation and Research and treated one million patients with living cell therapies. Earlier in his career, Mr. MacKay spent 11 years at Novartis in senior leadership positions, culminating as Vice-President Transplantation & Immunology. Past activities include Chairman of the Board of MassBio, Chairman of the Board of the Alliance of Regenerative Medicine, and a member of the advisory council to the Health Policy Commission for Massachusetts.
Franklin Berger, Director
Mr. Berger has more than 25 years of experience in capital markets and financial analysis. He serves on the Board of Directors of BELLUS Health sold to GSK, ESSA Pharma, Kezar Life Sciences, Atreca, Rain Therapeutics and Atea Pharmaceuticals. Mr. Berger served as a senior portfolio manager at Sectoral Asset Management; additionally, he was co-founder, co-PM on the small-cap focused NEMO Fund at Sectoral, from 2007 through June 2008. Previously, he was Managing Director, Equity Research and Senior Biotechnology Analyst for J. P. Morgan Securities from 1998 to 2003. During his time at J.P. Morgan, he was involved with the issuance of more than $12 billion in biotechnology company equity or equity-linked securities, including the Genentech initial public offering, the largest biotechnology financing to date. From 1997 to 1998, he served as a Director, Equity Research and Senior Biotechnology Analyst for Salomon Smith Barney and from 1991 to 1997, he served as a sell-side analyst for Josephthal & Co. The Wall Street Journal
- 62 -
selected Mr. Berger as the No. 1 ranked biotechnology analyst in its All-Star Analyst Survey in 1997 and was ranked No. 2 in the WSJ’s 2000 Survey. In 2002, Institutional Investor Magazine ranked him on J. P. Morgan’s 3rd-placed All-Star Research Team. Mr. Berger received a B.A. in International Relations, an M.A. in International Economics from Johns Hopkins University and an MBA from Harvard University. He serves on the Council of Rockefeller University and was a Founding Fellow of the Biotechnology Study Center at New York University School of Medicine.
Brian Bloom, Director
Mr. Bloom is a co-founder of Bloom Burton & Co. and serves as the firm’s Chairman and Chief Executive Officer. He serves on the Board of Directors of Satellos Bioscience and Appili Therapeutics. Mr. Bloom was formerly the Chairman of the Board of Grey Wolf Animal Health and Triumvira Immunologics, a member of the Life Sciences Advisory Board at the National Research Council Canada, the Dean’s Advisory Board at McMaster University and on the Board of Directors of BIOTECanada, the Baycrest Foundation and Qing Bile Therapeutics. Before co-founding Bloom Burton in 2008, he spent six years at an independent investment dealer in the healthcare and biotechnology institutional sales and equity research groups. Mr. Bloom started his career at New York-based investment banking firms SCO Financial Group and Molecular Securities. He is the proud recipient of the McMaster University 2017 Distinguished Alumni Award in Science and the co-recipient of the 2023 Life Sciences Ontario Community Service Award. Mr. Bloom received an Honours Bachelor of Science in Biochemistry from McMaster University and subsequently studied at the Mount Sinai Graduate School for Biological Sciences of New York University, with a focus in molecular endocrinology and biophysics.
Adam Mostafa, Director
Mr. Mostafa is an accomplished financial leader in strategic and financial planning in the pharmaceutical industry. Currently, Mr. Mostafa is the Chief Financial Officer of Sitryx Pharmaceuticals, a Senior Advisor to Cumberland Pharmaceuticals (NASDAQ: CPIX) and also sits on the Board of Precision Biologics. Previously, he was the CFO and Head of Business Development at X4 Pharmaceuticals (NASDAQ: XFOR), working closely with the Board on all strategic and financial matters. Notably this included the company’s Nasdaq public listing, multiple follow-on financings and strategic partnerships. Mr. Mostafa has served as CFO and Head of Business Development at Abpro Corporation, a biotechnology company focused on antibody therapeutics. Previously, Mr. Mostafa was a Managing Director in the healthcare investment banking group at Cantor Fitzgerald, and a senior banker in the healthcare investment banking group at Needham & Company. Mr. Mostafa has also held positions of vice president in the investment banking group at CRT Capital Group, portfolio management associate in the global stock selection group at AQR Capital, and analyst in the healthcare investment banking group at Salomon Smith Barney. Mr. Mostafa holds an A.B. in economics from Brown University.
Stephanie Brown, Director
Ms. Brown is a seasoned executive leader with over 30 years of experience in the bio-pharma industry, holding key leadership roles in North America, Europe, and Asia. Known for her strategic agility and deep operational expertise, Ms. Brown has driven successful business transformations and product launches across diverse therapeutic areas, from breakthrough biologics to small molecules. She has served in executive roles at top pharmaceutical companies, including Merck, Genentech, Biogen, Takeda, and Novartis, where she managed high-value product portfolios and led launches for multiple specialty and rare disease treatments.
Ms. Brown is Past-President of North America for Santhera Pharmaceuticals, where she oversaw strategic operations and corporate restructuring. Prior to Santhera, she led the Rare Diseases business at Ipsen Biopharmaceutical North America, managing commercialization strategies and profit growth for in-line and launch brands. Currently, she serves on the Board of Directors for Resilia, Inc., and has previously held board roles with ObsEva and the Biotechnology Innovation Organization (BIO). Ms. Brown holds a B.S. in chemistry with biology from Mount Allison University and an MBA from Edinburgh Business School, Heriot-Watt University.
Selwyn Ho, Director
Dr. Selwyn Ho is a seasoned Pharma and Biotech leader with extensive experience in commercial strategy, corporate leadership, financing, and board governance. As a trained medical doctor with a track record spanning multiple commercial, development and medical roles, he currently serves as CEO of Signadori Bio, a Sofinnova backed, privately held oncology company, spun out of Gustave Roussy, the leading European Cancer Centre.
Previously, as CEO of Medigene AG, a publicly traded immuno-oncology company, he led the company’s strategic transformation, streamlining R&D, securing key partnerships with BioNTech, Regeneron, and WuXi Biologics, and successfully driving financing
- 63 -
rounds. Prior to this, as EVP & Chief Business Officer at Connect Biopharma, Dr. Ho played a key role in raising $135 million in private financing followed by a further $220 million through the company’s NASDAQ IPO. His leadership experience includes senior roles at Dermira (Eli Lilly), UCB, Allergan (AbbVie), Novartis, and AstraZeneca, providing him with deep expertise in product launch and commercialization, market access, and global product strategy across immunology, oncology, and dermatology.
Dr. Ho also brings strong board and advisory experience, currently serving as a Non-Executive Director at Peptomyc S.L., as a Strategic Advisor to Antengene Corporation (6996.HK), an Advisor to Sofinnova Partners (Biovelocita Strategy) and as an Executive-in-Residence at New Rhein Healthcare Investors. Dr. Ho received his medical degree (MB, BS) and Bachelor of Science (BSc) in Pharmacology from Imperial College, University of London, UK, and post-graduate qualifications (Dip Pharm Med) in Pharmaceutical Medicine from the Faculty of Pharmaceutical Physicians, Royal College of Physicians, UK.
Iris Loew-Friedrich, Director
Dr. Iris Loew-Friedrich is a seasoned biotech and pharmaceutical executive with extensive experience in global drug development, regulatory affairs, corporate strategy, and board governance. In June 2024, Dr. Loew-Friedrich concluded her 20+ year tenure at UCB S.A, where she was most recently Executive Vice President, Chief Medical Officer, and Head of Development, leading global clinical development, regulatory affairs, medical affairs, safety/pharmacovigilance, and data strategy. Under her leadership, UCB successfully developed and secured approvals for multiple therapies, including Cimzia, Briviact, Evenity, Rystiggo, Zilbrysq, and Bimzelx across the US, EU, and international markets. She also played a pivotal role in lifecycle management, new formulations, and expanded indications across UCB’s portfolio.
Previously, Dr. Loew-Friedrich held executive roles at Schwarz Pharma, BASF Pharma, and Hoechst/Aventis, where she oversaw global R&D organizations, led successful regulatory approvals, and managed key industry partnerships. Notably, she contributed to the development and commercialization of Humira, Arava, Actonel, Vimpat, and Neupro. She received her medical license from Johann Wolfgang Goethe University, Frankfurt, Germany, and her Ph.D. following a research assignment at the Max-Planck-Institute for Biophysics. Dr. Loew-Friedrich served as the staff physician at the Department for Internal Medicine at the Frankfurt University Medical School, with sub-specialties in immunology, nephrology, and transplantation medicine. She is board-certified in Internal Medicine and holds a professorship at Frankfurt University Medical School.
She has extensive board experience, currently serving as Chair of the Supervisory Board at Evotec SE (until June 2026), Chair of the Board at Celosia Therapeutics, and a Non-Executive Director at Fresenius SE & Co. KGaA, Swedish Orphan Biovitrum AB and Financière de Tubize. Additionally, she has played key roles in scientific advisory boards and nonprofit organizations, including Helmholtz Health, Pierre Fabre S.A., and Fondazione Telethon, with a focus on oncology, immunology, and rare diseases.
Mark Nawacki, Director
Mr. Mark Nawacki is the co-founder and former President and CEO of Searchlight Pharma. Mr. Nawacki brings almost three decades of experience across pharmaceuticals, corporate development, and mergers and acquisitions. He co-founded Searchlight Pharma in 2015 and served as President and CEO from its inception till May 2024, guiding the organization’s expansion through acquisitions, in-licensing, and commercial growth into one of the three largest specialty pharmaceutical businesses in Canada. Upon Searchlight’s sale to Apotex in June 2024, Mark was appointed to the Apotex executive leadership team and continued to lead the innovative branded business as President until March 2026. His earlier career includes senior corporate development roles at Paladin Labs and board positions with healthcare companies.
Mr. Nawacki holds a BA in International Relations and Russian and East European Studies from the University of Toronto (Trinity College) and an MBA from the University of Toronto, and is a Fellow of the Canadian Chartered Professional Accountants of Ontario (FCPA, FCA). He previously served on the Board of Trustees of the Licensing Executives Society (USA & Canada) and is a former President and Board Member of the Canadian Healthcare Licensing Association.
Michael Rudnicki, OC, PhD, FRS, FRSC, Chief Discovery Officer
An acknowledged world thought leader and scientific authority on muscle stem cell function and their role in muscle regeneration, Dr. Rudnicki has maintained a continuous record of foundational findings and discoveries spanning a 40-year research career. In a landmark 2007 Cell paper, Dr. Rudnicki was the first to define and characterize a subpopulation of satellite cells as bona fide multipotent muscle stem cells in muscle tissue capable of both self-renewal and regeneration. His work transformed the field’s understanding of the
- 64 -
nature and role muscle stem cells play in the lifelong repair and growth processes of skeletal muscle, the body’s largest organ. Currently, Dr. Rudnicki is the Scientific Director of the Canadian Stem Cell Network, Director of the Sprott Centre for Stem Cell Research at the Ottawa Hospital Research Institute, a Professor in the Faculty of Medicine at the University of Ottawa. He has published more than 275 scientific publications and authored 17 patents. Dr. Rudnicki’s contributions have been recognized with numerous honours including being awarded a Tier 1 Canada Research Chair, a Fellow of both the Royal Society London and the Royal Society of Canada, a past International Research Scholar of the Howard Hughes Medical Institute, and an Officer of the Order of Canada. Dr. Rudnicki received his Ph.D. at the University of Ottawa and trained at the postdoctoral level at the Massachusetts Institute of Technology at the Whitehead Institute.
Dr. Rudnicki works part-time (approximately 20%) as a contractor to Satellos.
Phil Lambert, PhD, Chief Scientific Officer
Dr. Lambert is a neuroscientist by training and a high-performing preclinical executive with more than 25 years of drug discovery and development expertise in the areas of neuroscience, ocular, metabolic, inflammatory and orphan diseases. He has supported the advancement of more than 20 therapeutics, both small and large molecules, into clinical development by providing necessary preclinical data for multiple diseases. Dr. Lambert brings with him his accomplishments as a well published researcher, his unique senior leadership positions in academia, biotechnology, non-profit, large pharmaceutical and contract research organizations. As a serial entrepreneur, he was a member of the management team which conducted the due diligence and sale of Sirtris to GSK in 2008. Additionally, Dr. Lambert co-founded, effectively grew, and sold VivoPath, a contract research organization to Charles River Laboratories in 2014. Dr. Lambert brings his extensive leadership experience to Satellos, having held positions at Centogene, Life Biosciences, Sirtris Pharmaceuticals, Forum Pharmaceuticals, ALS Therapy Development Institute, Regeneron Pharmaceuticals, GSK and Parke Davis. He was a faculty member in the Department of Psychiatry at Emory University School of Medicine. Dr. Lambert received his Ph.D. in neuroendocrinology from Imperial College, University of London.
Elizabeth Williams, CPA, CA, Chief Financial Officer
Ms. Williams has more than 20 years of experience in biotech, working with publicly listed entities in both Canada and the United States. Prior to joining Satellos, Ms. Williams was CFO of Medicenna (Nasdaq: MDNA), where she was responsible for all financial, legal, and investor relations functions and led the graduation of the company from the TSXV to the TSX main board and subsequently the Nasdaq. Previous to Medicenna, she was Vice President of Finance and Administration at Aptose Biosciences Inc. (Nasdaq: APTO), a biotechnology company listed on both the TSX and the Nasdaq. While at Aptose, Ms. Williams held several positions, including Acting CFO, and was responsible for a broad range of activities including financings, financial reporting, and regulatory compliance. She serves as Director and Chair of the Audit Committee of Triumvira Immunologics Inc. and as a Director and Chair of the Audit Committee of the Stem Cell Network. Ms. Williams is a Chartered Professional Accountant (CPA) and Chartered Accountant (CA).
Wildon Farwell, M.D., Chief Medical Officer
Dr. Wildon Farwell has over a decade of leadership experience in clinical development and medical affairs, with deep expertise in neuromuscular and rare diseases. He most recently served as chief medical officer and later as medical advisor at Dyne Therapeutics, where he established and led the development organization. At Dyne, he oversaw protocol design and regulatory strategy for Duchenne muscular dystrophy (DMD) and myotonic dystrophy type 1 (DM1) programs and guided the execution of multiple potentially registrational clinical trials.
Prior to Dyne, Dr. Farwell spent ten years at Biogen, advancing through roles of increasing responsibility to become vice president of Late-Stage Clinical Development and global medical head of Neuromuscular Diseases. At Biogen, he led the development and lifecycle management of SPINRAZA®, the first FDA-approved treatment for spinal muscular atrophy, and initiated late-stage development of QALSODY®, an investigational therapy for amyotrophic lateral sclerosis (ALS). He also played a key role in biomarker strategy and pharmacovigilance across a range of therapeutic areas.
Before entering the biotechnology industry, Dr. Farwell was an assistant professor of Medicine at Harvard Medical School and practiced at both Brigham and Women’s Hospital and the VA Boston Healthcare System. He earned his medical degree from the University of Missouri School of Medicine and holds a Master of Public Health in Clinical Effectiveness from the Harvard T.H. Chan School of Public Health.
- 65 -
Cease Trade Orders
To the best of our knowledge, no director or executive officer of the Company, is, or within the ten years prior to the date hereof, has been, a director, chief executive officer or chief financial officer that: (i) while that person was acting in that capacity was the subject of a cease trade order or similar order or an order that denied the other issuer access to any exemptions under Canadian securities legislation, that was in effect for a period of more than thirty consecutive days or, (ii) after that person ceased to act in that capacity, was the subject of a cease trade order or similar order, or an order that denied the other issuer access to any exemptions under Canadian securities legislation, that was in effect for a period of more than thirty consecutive days and which resulted from an event that occurred while that person was acting in that capacity.
Penalties or Sanctions
To the best of our knowledge, no director or executive officer of the Company, or a shareholder holding a sufficient number of shares of the Company to affect materially the control of the Company, has been subject to any penalties or sanctions imposed by a court relating to Canadian securities legislation or by a Canadian securities regulatory authority or has entered into a settlement agreement with a Canadian securities authority, or any other penalties or sanctions imposed by a court or regulatory body that would likely be considered important to a reasonable investor in making an investment decision.
Personal Bankruptcies
Other than as described below, to the best of our knowledge, no director or executive officer of the Company, or a shareholder holding a sufficient number of shares of the Company to affect materially the control of the Company, (i) has, during the ten years prior to the date hereof, been a director or executive officer of any company that, while that person was acting in that capacity, became bankrupt, made a proposal under any legislation relating to bankruptcy or insolvency, or been subject to or instituted any proceedings, arrangement or compromise with creditors, or had a receiver, receiver manager or trustee appointed to hold his, her or its assets or (ii) has, during the ten years prior to the date hereof, become bankrupt, made a proposal under any legislation relating to bankruptcy or insolvency, or been subject to or instituted any proceedings, arrangement or compromise with creditors, or had a receiver, receiver manager or trustee appointed to hold his, her or its assets.
Stephanie Brown was previously a director of ObsEva SA (“ObsEva”). She resigned as a director in June 2023 and has had no further involvement with ObsEva since that date. On January 29, 2024, the Tribunal de première instance of Geneva granted a temporary moratorium (sursis provisoire) to ObsEva for a period of four months ending May 29, 2024 and appointed a commissioner (commissaire) to supervise the company’s activities during the process. On February 28, 2024 ObsEva announced that it would wind-down its operations with the termination of all of its employees and notified the SIX Swiss Exchange’s listing authority that there was a substantial risk that it would be unable to have audited financial statements for 2023 prepared and that it is as a result likely not going to be able to satisfy the requirements for maintaining its listing on SIX.
Selwyn Ho is a director and officer of Medigene AG which filed for insolvency on April 25, 2025. On April 7, 2025, Medigene announced it had filed an application for the opening of insolvency proceedings with the competent local court in Munich on April 16, 2025. On April 25, 2025, attorney Axel W. Bierbach from the law firm Müller-Heydenreich Bierbach & Kollegen was appointed preliminary insolvency administrator. Medigene AG’s research operations are currently being continued in full.
Conflicts of Interest
The Company’s directors and officers may serve as directors or officers of other companies or have significant shareholdings in other companies and, to the extent that such other companies may participate in ventures in which the Company may participate, the directors and officers of the Company may have a conflict of interest in negotiating and concluding terms respecting the extent of such participation. In the event that such a conflict of interest arises at a meeting of the Company’s directors, a director who has such a conflict will disclose his interest in the matter and abstain from voting for or against the approval of such participation or such terms. In accordance with the laws of Canada, the directors of the Company are required to act honestly, in good faith and in the best interests of the Company.
The directors and officers of the Company are aware of the existence of laws governing the accountability of directors and officers for corporate opportunity and requiring disclosures by the directors of conflicts of interest and the Company will rely upon such laws in respect of any directors’ and officers’ conflicts of interest or in respect of any breaches of duty by any of its directors and officers. All
- 66 -
such conflicts will be disclosed by such directors or officers in accordance with the laws of Canada and they shall govern themselves in respect thereof to the best of their ability in accordance with the obligations imposed upon them by law. Other than as disclosed under the heading “Interest of Management and Others in Material Transactions” below, the directors and officers of the Company are not aware of any such conflicts of interests.
COMMITTEES OF THE BOARD
The following is a description of the current committees of the Board:
Audit Committee
The mandate of the Audit Committee is to assist the Board in fulfilling its oversight responsibilities with respect to the financial affairs of the Company, including responsibility to:
| ● | oversee the integrity of the Company’s financial statements and financial reporting process, audit process, internal accounting controls and procedures and compliance with related legal and accounting principles; |
| ● | oversee the qualifications and independence of the external auditor; |
| ● | oversee the work of the Company’s financial management, internal audit function (if any) and external auditor in these areas; and |
| ● | provide an open avenue of communication between the external auditor, the internal auditors (if any), the Board and the Company’s management. |
In addition, the Committee shall prepare, if required, an audit committee report for inclusion in the information circular prepared in connection with the Company’s annual meeting of shareholders, in accordance with applicable rules and regulations. The members of the Audit Committee are Mr. Adam Mostafa, (Chair), Ms. Stephanie Brown, and Dr. Selwyn Ho.
Compensation Committee
The mandate of the Compensation Committee includes assisting the Board in discharging its responsibilities relating to compensation of the Company’s directors and executives, oversight of the Company’s overall compensation structure, policies and programs, review of the Company’s processes and procedures for the consideration and determination of director and executive compensation; and producing a report on executive compensation for inclusion in the Company’s information circular as required by applicable rules and regulations. The primary objective of the Committee is to develop and implement compensation policies and plans that ensure the attraction and retention of key management personnel, the motivation of management to achieve the Company’s corporate goals and strategies, and the alignment of the interests of management with the long-term interests of the Company’s shareholders. The members of the Compensation Committee are Mr. Geoff Mackay (Chair), Mr. Adam Mostafa and Ms. Stephanie Brown.
Nominating and Corporate Governance Committee
The mandate of the Nominating and Corporate Governance Committee is to support the Board of Directors in exercising its corporate governance functions, including identifying individuals qualified to become Board members, and recommend that the Board select the director nominees for the next annual meeting of shareholders; and to develop and recommend to the Board the corporate governance guidelines and processes applicable to the Company, review these guidelines and processes at least annually and recommend changes to the Board. The members of the Nominating and Corporate Governance Committee are Mr. Franklin Berger (Chair), Dr. Iris Loew-Friedrich and Dr. Selwyn Ho.
- 67 -
Audit Committee’s Charter
The Charter of the Audit Committee is attached hereto as Schedule “A”.
Composition of the Audit Committee
The Audit Committee consists of Adam Mostafa, Dr. Selwyn Ho and Stephanie Brown. All three members of the Audit Committee are considered “financially literate” and “independent”, as defined in National Instrument 52-110 – Audit Committees (“NI 52-110”).
Relevant Education and Experience
Each member of the Audit Committee has the industry experience necessary to understand and analyze financial statements of the level of complexity of Satellos, as well as the understanding of internal controls and procedures necessary for financial reporting. The specific education and experience of each is set out under their respective names under “Directors and Officers – Biographical Information and Principal Occupations” above.
Audit Committee Oversight
At no time since the commencement of the Company’s most recently completed financial year was a recommendation of the Audit Committee to nominate or compensate an external auditor not adopted by the Board.
Reliance on Certain Exemptions
At no time since the commencement of the Company’s most recently completed financial year has the Company relied on exemptions in relation to “De Minimus Non-Audit Services”, “Initial Public Offerings”, “Events Outside Control of Member”, “Death, Disability or Resignation of Audit Committee Member”, any exemption provided by Part 8 of NI 52-110, any exemption contained in Sections 3.3(2) or 3.6 of NI 52-110 or an exemption in relation to “Acquisition of Financial Literacy”.
Pre-Approval Policies and Procedures
The Audit Committee will pre-approve the appointment of external auditors for the engagement of non-audit services and may adopt specific pre-approval policies and procedures for such engagements, as described in the Audit Committee Charter which is attached hereto as Schedule “A”.
External Auditor Service Fees
On November 18, 2024, MNP LLP resigned at the request of the Corporation as auditor of the Corporation, and PricewaterhouseCoopers LLP (“PwC”) were appointed as the successor auditor. The following table sets forth the aggregate fees paid or payable for professional services rendered by the independent external auditors, PwC, for the fiscal years 2024 and 2025.
Financial Year | | | | Audit Related | | | | |
Ending | | Audit Fees | | Fees | | Tax Fees | | All Other Fees |
December 31, 2025 |
| Cdn$482,784 |
| Cdn$42,800 |
| Nil |
| Nil |
December 31, 2024 |
| Cdn$278,200 |
| Nil |
| Nil |
| Nil |
“Audit Fees” refers to the aggregate fees billed by the Company’s external auditors for audit services including interim reviews. “Audit Related Fees” refers to aggregate fees billed for assurance and related services by the Company’s external auditors that are reasonably related to the performance of the audit or review of the Company’s financial statements and not reported under Audit Fees, including the provision of comfort letters and consents, the consultation concerning financial accounting and reporting of specific issues and the review of documents filed with regulatory authorities. “Tax Fees” includes fees for professional services rendered by the Company’s external auditors for tax compliance, tax advice and tax planning. “All Other Fees” includes all fees billed by the Company’s external auditors for services not covered in the other three categories.
- 68 -
Prior to PwC, the Company’s auditor was MNP LLP. Fees paid for services provided in the fiscal year 2025 and for fees paid or payable for services rendered in 2024 up to and until their resignation were as follows.
Financial Year | | | | Audit Related | | | | |
Ending | | Audit Fees | | Fees | | Tax Fees | | All Other Fees |
December 31, 2025 |
| Nil |
| Cdn$76,740 |
| Cdn$5,350 |
| Nil |
December 31, 2024 |
| Cdn$77,040 |
| Cdn$65,125 |
| Cdn$22,804 |
| Nil |
“Audit Fees” refers to the aggregate fees billed by the Company’s external auditors for audit services including interim reviews. “Audit Related Fees” refers to aggregate fees billed for assurance and related services by the Company’s external auditors that are reasonably related to the performance of the audit or review of the Company’s financial statements and not reported under Audit Fees, including the provision of comfort letters and consents, the consultation concerning financial accounting and reporting of specific issues and the review of documents filed with regulatory authorities. “Tax Fees” includes fees for professional services rendered by the Company’s external auditors for tax compliance, tax advice and tax planning. “All Other Fees” includes all fees billed by the Company’s external auditors for services not covered in the other three categories.
- 69 -
Frank Gleeson may be considered to be a “promoter” of the Company within the meaning of applicable Canadian securities laws (a “Promoter”). Mr. Gleeson currently owns, or exercises control or direction over, 331,365 Common Shares representing approximately 2.1% of the issued and outstanding Common Shares on a non-diluted basis. Mr. Gleeson receives compensation from the Company for his services as Chief Executive Officer of the Company and as of the date hereof, has been granted 516,306 options pursuant to the Company’s stock option plan. The statement as to the number of Common Shares beneficially owned, or over which a Promoter exercises control or direction, directly or indirectly, not being within the knowledge of the Company, has been obtained from the System for Electronic Disclosure by Insiders.
- 70 -
LEGAL PROCEEDINGS AND REGULATORY ACTIONS
There are no pending or contemplated legal proceedings to which the Company is a party or of which any of our properties is the subject.
As of December 31, 2025, the Company is not subject to:
| (1) | any penalties or sanctions imposed against the Company by a court relating to securities legislation or by a securities regulatory authority during the financial year ended December 31, 2025; or |
| (2) | any other penalties or sanctions imposed by a court or regulatory body against the Company that would likely be considered important to a reasonable investor in making an investment decision; or |
| (3) | settlement agreements the Company entered into before a court relating to securities legislation or with a securities regulatory authority during the financial year ended December 31, 2025. |
The Company is unaware of any condition of default under any debt, regulatory, exchange related or other contractual obligation.
- 71 -
INTEREST OF MANAGEMENT AND OTHERS IN MATERIAL TRANSACTIONS
Except as described below, no director, officer or principal shareholder of the Company, or any associate or affiliate of any of the foregoing persons or entities, has any direct or indirect material interest in any transaction within three years of the date of this Annual Information Form or in any proposed transaction of the Company that has materially affected or will materially affect the Company or any of our subsidiaries.
On May 17, 2023, the Company completed the May 2023 Equity Offering for gross proceeds of Cdn$55 million. BBSI acted as exclusive agent and book running manager and as compensation the Company issued 546,706 compensation warrants, paid approximately Cdn$3.3 million in commissions and reimbursed BBSI for Cdn$176 thousand in legal and related fees related to the May 2023 Equity Offering.
On December 20, 2024, the Company completed the December 2024 Public Offering for gross proceeds of US$40 million. BBSI acted as lead agent for the December 2024 Public Offering and in connection therewith received a portion of the agency fee (aggregate agency fee equal to 7.0% of the gross proceeds, less shares subscribed by insiders).
On February 9, 2026, the Company completed the 2026 Offering, for gross proceeds of US$57.2 million. BBSI acted as co-agent for the 2026 Offering and in connection therewith received a portion of the underwriters’ fee (aggregate underwriters’ fee equal to 7.0% of the gross proceeds). In addition, Franklin Berger, a director of the Company, purchased 24,750 Common Shares in the 2026 Offering.
- 72 -
The registrar and transfer agent for the Common Shares is Canada and in the United States is Computershare Investor Services Inc., 510 Burrard Street, 3rd Floor, Vancouver, British Columbia, V6C 3B9.
- 73 -
The following are the material contracts that the Company or a subsidiary of the Company have entered into during the financial year ended December 31, 2025, or prior thereto but still in effect and that are required to be filed under National Instrument 51-102 Continuous Disclosure Obligations:
| (1) | OHRI License, as amended. |
| (2) | Underwriting Agreement dated February 5, 2026 among the Company and Leerink Partners LLC, on behalf of a syndicate of underwriters, with respect to the 2026 Offering. See “General Development of the Business – Three Year History – Subsequent Events”. |
- 74 -
The Company’s independent registered public accounting firm is PricewaterhouseCoopers LLP, Chartered Professional Accountants, who issued a Report of Independent Registered Public Accounting Firm dated March 27, 2026 in respect of the Company’s consolidated financial statements as at December 31, 2025 and 2024 and for years then ended. PricewaterhouseCoopers LLP has advised the Company that they are independent with respect to the Company within the meaning of the relevant rules and related interpretations prescribed by the relevant professional bodies in Canada, including the Chartered Professional Accountants of Ontario CPA Code of Professional Conduct and any applicable legislation or regulations and the rules and regulations adopted by the SEC and Public Company Accounting Oversight Board (United States).
- 75 -
Additional information with respect to Satellos may be found on its SEDAR+ profile at www.sedarplus.com. Additional financial information is provided in the Company’s financial statements and Management’s Discussion and Analysis for the year ended December 31, 2025.
Additional information, including directors’ and officers’ remuneration and indebtedness, principal holders of the Company’s securities and securities authorized for issuance under equity compensation plans, if applicable, is contained in the Company’s management information circular dated May 12, 2025 for its annual general and special meeting of Shareholders held on June 18, 2025.
- 76 -
- A-1 -
SATELLOS BIOSCIENCE INC.
(the “Company”)
AUDIT COMMITTEE CHARTER
A.Purpose
The Audit Committee (the “Committee”) is a standing committee appointed by the board of directors (the “Board”) of the Company. The Committee is established to assist the Board in fulfilling its oversight responsibilities with respect to the financial affairs of the Company, including responsibility to:
| ● | oversee the integrity of the Company’s financial statements and financial reporting process, audit process, internal accounting controls and procedures and compliance with related legal and accounting principles; |
| ● | oversee the qualifications and independence of the external auditor; |
| ● | oversee the work of the Company’s financial management, internal audit function (if any) and external auditor in these areas; and |
| ● | provide an open avenue of communication between the external auditor, the internal auditors (if any), the Board and the Company’s management. |
In addition, the Committee shall prepare, if required, an audit committee report for inclusion in the information circular prepared in connection with the Company’s annual meeting of shareholders, in accordance with applicable rules and regulations.
The function of the Committee is oversight. It is not the duty or responsibility of the Committee or its members (i) to plan or conduct audits, (ii) to determine that the Company’s financial statements are complete and accurate and are in accordance with international financial reporting standards (“IFRS”) or (iii) to conduct other types of auditing or accounting reviews or similar procedures or investigations. The Committee members and its Chair are members of the Board, appointed to the Committee to provide broad oversight of the financial, risk and control-related activities of the Company, and are specifically not accountable or responsible for the day-to-day operation or performance of such activities. In particular, the member or members identified as audit committee financial experts, if any, shall not be accountable for giving professional opinions on the internal or external audit of the Company’s financial information.
Management is responsible for the preparation, presentation and integrity of the Company’s financial statements. Management is also responsible for ensuring that adequate systems of risk assessment and internal controls and procedures are designed and put in place in accordance with the accounting policies determined by the Committee to provide reasonable assurance that assets are safeguarded and transactions are properly authorized, recorded and reported and to assure the effectiveness and efficiency of operations, the reliability of financial reporting and compliance with accounting standards and with applicable laws and regulations. The internal auditor (if any) is responsible for monitoring and reporting on the adequacy and effectiveness of the system of internal controls. The external auditor is responsible for planning and carrying out an audit of the Company’s annual financial statements in accordance with IFRS to provide reasonable assurance that, among other things, such financial statements are in accordance with IFRS.
B.Procedures
| 1. | Composition – The Committee shall be comprised of at least three members. None of the members of the Committee shall be an officer or employee of the Company or any of its subsidiaries and the membership of the Committee shall satisfy the independence, experience and financial expertise requirements of any and all securities exchange(s) on which the securities of the Company are listed and posted for trading and all other applicable securities and other laws and none of the members shall have participated in the preparation of the financial statements of the Company or any current subsidiaries of the Company at any time over the past three years. |
| 2. | Appointment and Replacement of Committee Members – Any member of the Committee may be removed or replaced at any time by the Board and shall automatically cease to be a member of the Committee upon ceasing to be a director. The Board may fill vacancies on the Committee by appointing another director to the Committee. The Board shall fill any vacancy if the membership of the Committee is less than three directors or if the Committee does not have at least one member with accounting |
- A-2 -
| or related financial expertise. Whenever there is a vacancy on the Committee, the remaining members may exercise all its power as long as a quorum remains in office. Subject to the foregoing, the members of the Committee shall be appointed by the Board annually and each member of the Committee shall remain on the Committee until the next annual meeting of shareholders after his or her election or until his or her successor shall be duly elected and qualified. |
| 3. | Committee Chair – Unless a Chair of the Committee is designated by the full Board, the members of the Committee may designate a Chair by majority vote of the full Committee. The Chair of the Committee shall be responsible for leadership of the Committee, including preparing the agenda, presiding over the meetings, making committee assignments and reporting to the Board. |
| 4. | Conflicts of Interest – If a Committee member faces a potential or actual conflict of interest relating to a matter before the Committee, other than matters relating to the compensation of directors, that member shall be responsible for alerting the Chair of the Committee. If the Chair of the Committee faces a potential or actual conflict of interest, the Chair of the Committee shall advise the Chair of the Board. If the Chair of the Committee, or the Chair of the Board, as the case may be, concurs that a potential or actual conflict of interest exists, then the member faced with such conflict shall disclose to the Committee the member’s interest and shall not participate in consideration of the matter and shall not vote on the matter. |
| 5. | Compensation of Committee Members – The members of the Committee shall be entitled to receive such remuneration for acting as members of the Committee as the Board may from time to time determine. No member of the Committee shall receive from the Company or any of its affiliates any compensation other than the fees to which he or she is entitled as a director or a member of the Committee of the Board or any of its affiliates. |
| 6. | Meetings of the Committee |
| (a) | Procedures for Meetings – Subject to any applicable statutory or regulatory requirements, the articles and by-laws of the Company and the terms of this Charter, the time at which and place where the meetings of the Committee shall be held and the calling of Committee meetings and the procedure in all things at such meetings shall be determined by the Committee, provided that it is understood that the Committee may meet in person and by telephone or electronic means that permit all persons participating in the meeting to communicate simultaneously and instantaneously and that the Committee may act by means of a written resolution signed by all members entitled to vote on the matter. |
| (b) | Calling of Meetings – The Committee shall meet as often as it deems appropriate to discharge its responsibilities. Notice of the time and place of every meeting shall be given in writing, by any means of transmitted or recorded communication, including email, video conferences or other electronic means that produces a written copy, to each member of the Committee at least 24 hours prior to the time fixed for such meeting. However, a member may in any manner waive a notice of a meeting. Attendance of a member at a meeting constitutes a waiver of notice of the meeting, except where a member attends a meeting for the express purpose of objecting to the transaction of any business on the grounds that the meeting is not lawfully called. Whenever practicable, the agenda for the meeting and the meeting materials shall be provided to members before the Committee meeting in sufficient time to provide adequate opportunity for their review. |
| (c) | Quorum – A majority of the members of the Committee constitute a quorum for the transaction of Committee business. |
| (d) | Chair of Meetings – If the Chair of the Committee is not present at any meeting of the Committee, one of the other members of the Committee who is present shall be chosen by the Committee to preside at the meeting. |
| (e) | Secretary of Meeting – The Chair of the Committee shall designate a person who need not be a member of the Committee to act as secretary or, if the Chair of the Committee fails to designate such a person, the secretary of the Company shall be secretary of the Committee. The agenda of each Committee meeting will be prepared by the secretary of the Committee and, whenever reasonably practicable, circulated to each member prior to each meeting. |
| (f) | Separate Executive Meetings – The Committee shall meet at least once every year, and more often as warranted, with the Chief Executive Officer and such other officers of the Company as the Committee may determine to discuss any matters that the Committee or such individuals believes should be discussed privately. |
- A-3 -
| (g) | Minutes – Minutes of the proceedings of each Committee meeting shall be kept in minute books provided for that purpose. The minutes of Committee meetings shall accurately record the discussions of and decisions made by the Committee, including all recommendations to be made by the Committee to the Board and shall be distributed to all Committee members. |
C.AUDIT RESPONSIBILITIES OF THE COMMITTEE
Fundamental Powers
| 7. | Subject to any applicable statutory or regulatory requirements, the articles and by-laws of the Company and the terms of this Charter, the Committee shall have the following fundamental powers in addition to any powers set out in this Charter or otherwise specified by the Board from time to time: |
| (a) | Access – The Committee is entitled to full access to all books, records, facilities, and personnel of the Company and its subsidiaries. The Committee may require such officers, directors and employees of the Company and its subsidiaries and others as it may see fit from time to time to provide any information about the Company and its subsidiaries it may deem appropriate and to attend and assist at meetings of the Committee. |
| (b) | Delegation – The Committee may delegate from time to time to any person or committee of persons any of the Committee’s responsibilities that lawfully may be delegated. |
| (c) | Adoption of Policies and Procedures – The Committee may adopt policies and procedures for carrying out its responsibilities. |
The Committee shall have the ability to communicate directly with the external auditor and the Company’s internal auditor, if any.
The Committee has the authority, to the extent it deems necessary or appropriate, to retain independent legal, accounting or other advisors (“Advisors”). The Company will provide appropriate funding, as determined by the Committee, for payment of compensation to the external auditors for the purpose of rendering or issuing an audit report and to any Advisors employed by the Committee.
Selection and Oversight of the External Auditor
| 8. | The external auditor is ultimately accountable to the Committee and the Board as the representatives of the shareholders of the Company and shall report directly to the Committee and the Committee shall so instruct the external auditor. The Committee shall evaluate the performance of the external auditor and make recommendations to the Board on the appointment, reappointment or replacement of the external auditor of the Company to be proposed in the Company’s information circular for shareholder approval and shall have authority to terminate the external auditor. If a change in external auditor is proposed, the Committee shall review the reasons for the change and any other significant issues related to the change, including the response of the incumbent auditors, and enquire as to the qualifications of the proposed auditors before making its recommendation to the Board. |
| 9. | The Committee shall approve in advance the terms of engagement and the compensation to be paid by the Company to the external auditor with respect to the conduct of the annual audit. The Committee may approve policies and procedures for the pre-approval of services to be rendered by the external auditor, which policies and procedures shall include reasonable detail with respect to the services covered. All non-audit services to be provided to the Company or any of its affiliates by the external auditor or any of its affiliates which are not covered by pre-approval policies and procedures approved by the Committee shall be subject to pre-approval by the Committee. |
| 10. | The Committee shall review the independence of the external auditor and shall make recommendations to the Board on appropriate actions to be taken which the Committee deems necessary to protect and enhance the independence of the external auditor. In connection with such review, the Committee shall: |
- A-4 -
| (a) | actively engage in a dialogue with the external auditor about all relationships or services that may impact the objectivity and independence of the external auditor; |
| (b) | require that the external auditor submit to it on a periodic basis and, at least annually, a formal written statement delineating all relationships between the Company and its subsidiaries, on the one hand, and the external auditor and its affiliates, on the other hand; |
| (c) | consider whether there should be a regular rotation of the audit partners responsible for performing the audit and/or of the external audit firm itself; and |
| (d) | consider the auditor independence standards promulgated by applicable auditing regulatory and professional bodies. |
| 11. | The Committee shall consider whether to prohibit the external auditor and its affiliates from providing certain non-audit services to the Company and its affiliates. |
| 12. | The Committee shall require the external auditor to provide to the Committee, and the Committee shall review and discuss with the external auditor, all reports which the external auditor is required to provide to the Committee or the Board under rules, policies or practices of professional or regulatory bodies applicable to the external auditor, and any other reports which the Committee may require. |
| 13. | The Committee is responsible for resolving disagreements between management and the external auditor regarding financial reporting. |
Appointment and Oversight of Internal Auditors (If Any)
| 14. | The appointment, authority, budget, replacement or dismissal of the internal auditors, if any, shall be subject to prior review and approval by the Committee. When any such internal audit function is performed by employees of the Company or its subsidiaries, the Committee may delegate responsibility for approving the employment, term of employment, compensation and termination of employees engaged in such function other than the head of the Company’s internal audit function. |
| 15. | The Committee shall obtain from the internal auditors (if any), and shall review, summaries of the significant reports to management prepared by any such internal auditors (or the actual reports if requested by the Committee) and management’s responses to such reports. |
| 16. | The Committee shall, as it deems necessary, communicate with the internal auditors (if any) with respect to their reports and recommendations, the extent to which prior recommendations have been implemented and any other matters that such internal auditors bring to the attention of the Committee. The head of the internal audit function (if one exists) shall have unrestricted access to the Committee. |
| 17. | The Committee shall, annually or more frequently as it deems necessary, evaluate the internal auditors (if any), including their activities, organizational structure and qualifications and effectiveness. |
Oversight and Monitoring of Audits
| 18. | The Committee shall review with the external auditor, the internal auditors (if any) and management the audit function generally, the objectives, staffing, locations, co-ordination, reliance upon management and internal audit (if any) and general audit approach and scope of proposed audits of the financial statements of the Company and its subsidiaries, the overall audit plans, the responsibilities of management, the internal auditors (if any) and the external auditor, the audit procedures to be used and the timing and estimated budgets of the audits. |
| 19. | The Committee shall meet periodically as it deems necessary with the internal auditor (if any) to discuss the progress of their activities and any significant findings stemming from internal audits and any difficulties or disputes that arise with management and the adequacy of management’s responses in correcting audit-related deficiencies. |
- A-5 -
| 20. | The Committee shall discuss with the external auditor any difficulties or disputes that arose with management or the internal auditors (if any) during the course of the audit, any restrictions on the scope of activities or access to requested information and the adequacy of management’s responses in correcting audit-related deficiencies. |
| 21. | The Committee shall review with management the results of internal (if any) and external audits. |
| 22. | The Committee shall take such other reasonable steps as it may deem necessary to satisfy itself that the audit was conducted in a manner consistent with all applicable legal requirements and auditing standards of applicable professional or regulatory bodies. |
Oversight and Review of Accounting Principles and Practices
| 23. | The Committee shall, as it deems necessary, oversee, review and discuss with management, the external auditor and the internal auditors (if any): |
| (a) | the quality, appropriateness and acceptability of the Company’s accounting principles and practices and that of its subsidiaries used in its financial reporting, changes in the Company’s accounting principles or practices and that of its subsidiaries and the application of particular accounting principles and disclosure practices by management to new transactions or events; |
| (b) | all significant financial reporting issues and judgments made in connection with the preparation of the financial statements, including the effects of alternative methods within IFRS on the financial statements and any “second opinions” sought by management from any other auditor firm or advisor with respect to the accounting treatment of a particular item; |
| (c) | disagreements between management and the external auditor or the internal auditors (if any) regarding the application of any accounting principles or practices; |
| (d) | any material change to the Company’s auditing and accounting principles and practices or that of its subsidiaries as recommended by management, the external auditor or the internal auditors (if any) or which may result from proposed changes to applicable IFRS; |
| (e) | the effect of regulatory and accounting initiatives on the Company’s financial statements and other financial disclosures; |
| (f) | any reserves, accruals, provisions, estimates or management programs and policies, including factors that affect asset and liability carrying values and the timing of revenue and expense recognition, that may have a material effect upon the financial statements of the Company; |
| (g) | the use of special purpose entities and the business purpose and economic effect of off-balance sheet transactions, arrangements, obligations, guarantees and other relationships of the Company or its subsidiaries and their impact on the financial results of the Company; |
| (h) | any legal matter, claim or contingency that could have a significant impact on the financial statements, the Company’s compliance policies and that of its subsidiaries and any material reports, inquiries or other correspondence received from regulators or governmental agencies and the manner in which any such legal matter, claim or contingency has been disclosed in the Company’s financial statements; |
| (i) | the treatment for financial reporting purposes of any significant transactions which are not a normal part of the Company’s operations or those of its subsidiaries; |
| (j) | the use of any “pro forma” or “adjusted” information not in accordance with IFRS; and |
| (k) | management’s determination of goodwill impairment, if any, as required by applicable accounting standards. |
- A-6 -
Oversight and Monitoring of Internal Controls
| 24. | The Committee shall, as it deems necessary, exercise oversight of, review and discuss with management, the external auditor and the internal auditors (if any): |
| (a) | the adequacy and effectiveness of the Company’s internal accounting and financial controls and also of its subsidiaries and the recommendations of management, the external auditor and the internal auditors (if any) for the improvement of accounting practices and internal controls; |
| (b) | any significant deficiencies or material weaknesses in the internal control environment, including with respect to computerized information system controls and security; |
| (c) | any fraud that involves personnel who have a significant role in the Company’s internal control over financial reporting or that of its subsidiaries; and |
| (d) | management’s compliance with the Company’s processes, procedures and internal controls. |
Communications with Others
| 25. | The Committee shall establish and monitor procedures for the receipt and treatment of complaints received by the Company and its subsidiaries regarding accounting, internal accounting controls or audit matters and the anonymous submission by employees of concerns regarding questionable accounting or auditing matters and shall review periodically with management and the internal auditors (if any) these procedures and any significant complaints received. |
Oversight and Monitoring of the Company’s Financial Disclosures
| 26. | The Committee shall: |
| (a) | review with the external auditor and with management and shall recommend to the Board for approval the annual financial statements and the notes and Management’s Discussion and Analysis accompanying such financial statements and any related press release, the Company’s annual report and any financial information of the Company contained in any prospectus or information circular of the Company; and |
| (b) | review and recommend to the Board, as necessary, with the external auditor and with management each set of interim financial statements and the notes and Management’s Discussion and Analysis accompanying such financial statements and any related press release and any other disclosure documents or regulatory filings of the Company containing or accompanying financial information of the Company. |
Such reviews shall be conducted prior to the release of any summary of the financial results or the filing of such reports with applicable regulators.
| 27. | The Committee shall review the disclosure with respect to its pre-approval of audit and nonaudit services provided by the external auditor. |
Oversight of Finance and Financial Risk Matters
| 28. | Appointments of the key financial executives involved in the financial reporting process of the Company, including the Chief Financial Officer, shall require the prior review of the Committee. |
| 29. | The Committee shall receive and review (as applicable): |
| (a) | periodic reports on compliance with requirements regarding statutory deductions and remittances and, in the event of any non-compliance, the nature and extent of the non-compliance, the reasons therefor and management’s plan and timetable to correct any deficiencies; |
- A-7 -
| (b) | material policies and practices of the Company and its subsidiaries respecting cash management and material financing strategies or policies or proposed financing arrangements and objectives of the Company and its subsidiaries; and |
| (c) | material tax policies and tax planning initiatives, tax payments and reporting and any pending tax audits or assessments. |
| 30. | The Committee shall meet periodically with management to review and discuss the Company’s major financial risk exposures and the policy steps that management has taken to monitor and control such exposures, including the use of financial derivatives and hedging activities and the Company’s insurance programs. |
| 31. | The Committee shall receive and review the financial statements and other financial information of material subsidiaries of the Company and any auditor recommendations concerning such subsidiaries. |
| 32. | The Committee shall meet with management to review the process and systems in place for ensuring the reliability of public disclosure documents that contain audited and unaudited financial information and their effectiveness. |
Additional Responsibilities
| 33. | The Committee shall review and approve all related party transactions on an ongoing basis, including to the extent required by any and all securities exchange(s) on which the securities of the Company are listed and posted for trading. |
| 34. | The Committee shall review and/or approve any other matter specifically delegated to the Committee by the Board and undertake on behalf of the Board such other activities as may be necessary or desirable to assist the Board in fulfilling its oversight responsibilities with respect to financial reporting and the Company’s financial obligations. |
D.THE CHARTER
| 35. | The Committee shall review and reassess the adequacy of this Charter periodically as it deems appropriate and recommend changes to the Board. The performance of the Committee shall be evaluated with reference to this Charter annually or otherwise periodically as deemed appropriate by the Board. |
Adopted on January 30, 2024, last reviewed March 26, 2026
- A-8 -