liquid formulations of colchicine for the prophylactic treatment of gout (a painful arthritic disorder) in adult humans in the United States and (2) an exclusive, transferable license to use the trademark “GLOPERBA”. GLOPERBA is an FDA-approved, oral medication for the treatment of gout in adults and we are planning to commercialize GLOPERBA in 2023 and believe we are well-positioned to market and distribute the product.
Our Marketed Product and Innovative Non-Opioid Pipeline
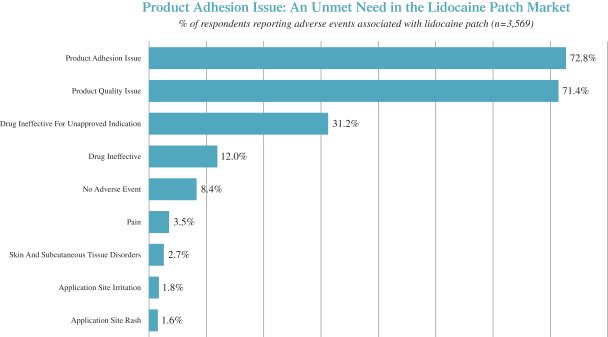
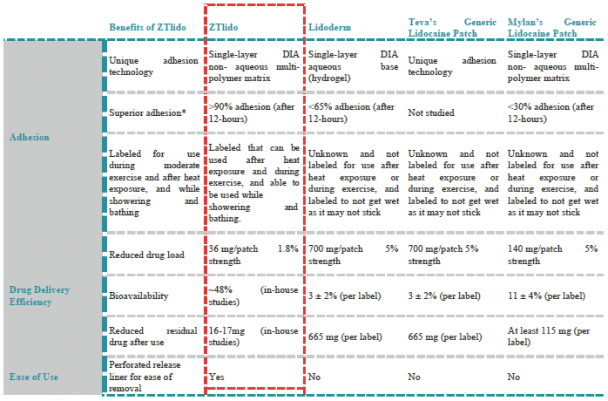
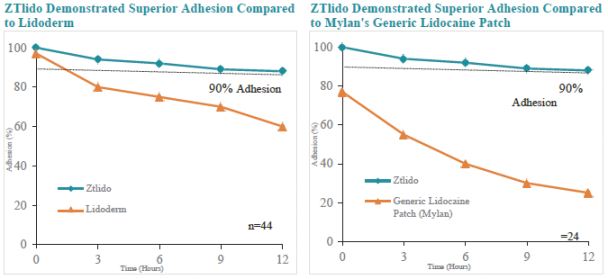
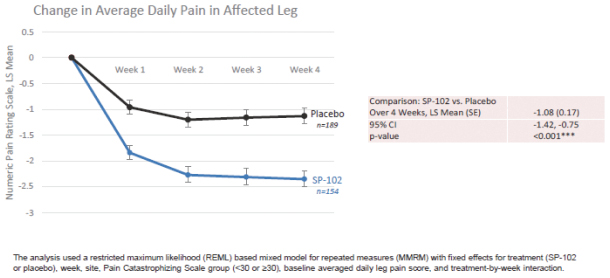
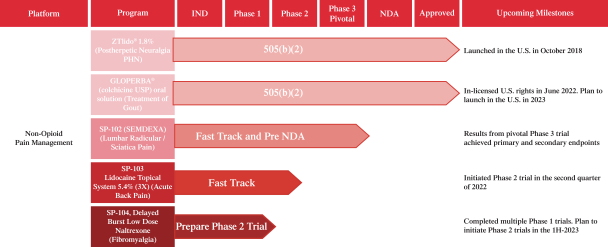
We are focused on developing and commercializing innovative non-opioid therapies that will provide safe, substantial and localized pain relief for large market opportunities. The following chart illustrates our current commercial product and novel product candidates, for which we have worldwide commercialization rights, except with respect to Japan for ZTlido and SP-103.
Our principal executive offices are located at 960 San Antonio Road, Palo Alto, California 94303, and our telephone number is (650) 516-4310. For additional information about us, please see the sections of this prospectus titled “Business” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
See the section titled “Risk Factors” in this prospectus for a discussion of some of the risks related to the execution of our business strategy.
On November 10, 2022 (the “Closing Date”), Vickers consummated the previously announced Business Combination pursuant to the terms of the Merger Agreement, by and among Vickers, Merger Sub and Legacy Scilex.
Pursuant to the Merger Agreement, (i) prior to the Closing, Vickers changed its jurisdiction of incorporation by deregistrating as a Cayman Islands exempted company and continuing and domesticating as a corporation incorporated under the laws of the State of Delaware (the “Domestication”) and (ii) at the Closing, and following the Domestication, Merger Sub merged with and into Legacy Scilex (the “Merger”), with Legacy Scilex as the surviving company in the Merger, and, after giving effect to such Merger, Legacy Scilex became a wholly owned subsidiary of Vickers. The Merger was approved by Vickers’s shareholders at a meeting held on November 9, 2022. In connection with the Business Combination, Vickers changed its name to “Scilex Holding Company.”
2