As filed with the United States Securities and Exchange Commission on September 28, 2021.
Registration No. 333-258879
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
___________________
AMENDMENT NO. 1
TO
FORM F-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
___________________
Incannex Healthcare Limited
(Exact name of registrant as specified in its charter)
___________________
|
Australia |
2834 |
Not Applicable |
||
|
(State or other jurisdiction of |
(Primary Standard Industrial |
(I.R.S. Employer |
Incannex Healthcare Limited
Suite 15, Level 12, 401 Docklands Drive
Docklands 3008, Victoria
Australia
+ 61 409 840 786
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
___________________
Vcorp Services, LLC
25 Robert Pitt Drive, Suite 204
Monsey, New York 10952
+1 888 528 2677
(Name, address, including zip code, and telephone number, including area code, of agent for service)
___________________
Copies to:
|
Andrew Reilly |
Jonathan Zimmerman |
___________________
Approximate date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act, check the following box: ☐
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act.
Emerging growth company ☒
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
Calculation of Registration Fee
|
Title of Each Class of Securities to be Registered |
Proposed |
Amount of |
||||
|
Ordinary shares, no par value(1)(3) |
$ |
25,000,000 |
$ |
2,730 |
||
|
Underwriter’s warrants(2) |
$ |
1,875,000 |
$ |
205 |
||
|
Ordinary shares issuable upon exercise of the Underwriter’s warrants(4) |
|
— |
|
— |
||
|
Total |
$ |
26,875,000 |
$ |
2,935 |
||
____________
(1) All ordinary shares in the offering will be in the form of American Depositary Shares, or ADSs, with each ADS representing 50 ordinary shares. ADSs issuable upon deposit of the ordinary shares registered hereby are being registered pursuant to a separate registration statement on Form F-6.
(2) We have calculated the proposed maximum aggregate offering price of the ordinary shares underlying the underwriter’s warrants to purchase up to 7.5% of the amount of securities sold in this offering by assuming that (i) 2.5% of such warrants are exercisable at a price per share equal to 120% of the public offering price of the ADSs sold in this offering, (ii) 2.5% of such warrants are exercisable at a price per share equal to 135% of the public offering price of the ADSs sold in this offering, (iii) 2.5% of such warrants are exercisable at a price per share equal to 150% of the public offering price of the ADSs sold in this offering. All ordinary shares will be in the form of ADSs, with each ADS representing 50 ordinary shares. ADSs issuable upon deposit of the ordinary shares registered hereby are being registered pursuant to a separate registration statement on Form F-6.
(3) Includes ordinary shares (which may be in the form of ADSs) that the underwriter has an option to purchase. See “Underwriting.”
(4) No additional registration fee is payable pursuant to Rule 457(i) under the Securities Act.
(5) Estimated solely for the purpose of computing the amount of the registration fee pursuant to Rule 457(o) under the Securities Act of 1933, as amended, based on an estimate of the proposed maximum offering price.
(6) The registrant previously paid $2,935 of the registration fee in connection with the filing of its initial registration statement on August 17, 2021.
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and we are not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to completion, dated September 28, 2021
PRELIMINARY PROSPECTUS

American Depositary Shares
representing Ordinary Shares
$ per American Depositary Share
___________________
We are offering American depositary shares, or ADSs, in the United States, representing ordinary shares of Incannex Healthcare Limited (“Incannex” or the “Company”). Each ADS represents 50 ordinary shares, no par value, deposited with Deutsche Bank Trust Company Americas, as depositary.
The offering is being underwritten on a firm commitment basis. We have granted the underwriter an option to buy up to an additional ADSs to cover over-allotments. The underwriter may exercise this option at any time and from time to time during the 30-day period from the date of this prospectus.
Prior to this offering, there has been no public market for the ADSs. We have applied to list the ADSs on the Nasdaq Capital Market under the symbol “IXHL”.
Our ordinary shares are listed on the Australian Securities Exchange under the symbol “IHL.” On September 27, 2021, the last reported sale price of our ordinary shares on the Australian Securities Exchange was A$0.355 per ordinary share, equivalent to a price of US$12.93 per ADS, after giving effect to the Australian dollar/U.S. dollar exchange rate of A$1.00 to US$0.7284 (as published by the Reserve Bank of Australia as of September 27, 2021), and an ADS-to-ordinary share ratio of 1 to 50. For the purposes of this preliminary prospectus, the estimated initial public offering price is US$12.93 per ADS.
The final offering price per ADS in U.S. dollars will be determined through negotiations between us and the representatives of the underwriter and will be based, in part, on prevailing market prices of our ordinary shares on the Australian Securities Exchange, after taking into account market conditions and other factors. For a discussion of the other factors considered in determining the final offering price per ADS, see “Underwriting.”
|
No Exercise of |
Full Exercise of |
|||||||||||
|
Per Share |
Total |
Per Share |
Total |
|||||||||
|
Public offering price |
$ |
$ |
$ |
$ |
||||||||
|
Underwriting discounts and commissions(1) |
$ |
$ |
$ |
$ |
||||||||
|
Proceeds to us, before expenses |
$ |
$ |
$ |
$ |
||||||||
____________
(1) In addition, we have agreed to reimburse the underwriter for certain expenses. See “Underwriting” on page 117 of this prospectus for additional information.
Investing in our securities involves a high degree of risk. See the section entitled “Risk Factors” appearing on pages 10 of this prospectus and elsewhere in this prospectus and the accompanying base prospectus for a discussion of information that should be considered in connection with an investment in our securities.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The underwriter expects to deliver the ADSs to purchasers on or about , 2021 through the book-entry facilities of The Depository Trust Company.
___________________
Roth Capital Partners
The date of this prospectus is , 2021
|
Page |
||
|
1 |
||
|
6 |
||
|
8 |
||
|
10 |
||
|
35 |
||
|
36 |
||
|
37 |
||
|
38 |
||
|
39 |
||
|
41 |
||
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
42 |
|
|
49 |
||
|
86 |
||
|
95 |
||
|
96 |
||
|
97 |
||
|
100 |
||
|
109 |
||
|
Material United States Federal Income and Australian Tax Considerations |
110 |
|
|
116 |
||
|
117 |
||
|
122 |
||
|
123 |
||
|
123 |
||
|
123 |
We are responsible for the information contained in this prospectus and any free writing prospectus we prepare or authorize. We and the underwriter have not authorized anyone to provide you with different information. We and the underwriter take no responsibility for, and can provide no assurance as to the reliability of, any other information others may give you. We are not, and the underwriter are not, making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted. You should not assume that the information contained in this prospectus is accurate as of any date other than its date.
For investors outside the United States: neither we nor any of the underwriter have done anything that would permit this offering or possession or distribution of this prospectus or any free writing prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus or any free writing prospectus must inform themselves about, and observe any restrictions relating to, the offering of the ADSs and the distribution of this prospectus and any free writing prospectus outside the United States.
We are incorporated under the laws of Australia, and a majority of our outstanding ordinary shares are owned by non-U.S. residents. Under the rules of the U.S. Securities and Exchange Commission, or the SEC, we are eligible for treatment as a “foreign private issuer.” As a foreign private issuer, we will not be required to file periodic reports and financial statements with the SEC as frequently or as promptly as domestic registrants whose securities are registered under the Securities Exchange Act of 1934, as amended, or the Exchange Act.
Our reporting and functional currency is the Australian dollar, and our financial statements included elsewhere in this prospectus are presented in Australian dollars. The consolidated financial statements and related notes included elsewhere in this prospectus have been prepared under the International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB, which differs in certain significant respects from U.S. Generally Accepted Accounting Principles, or GAAP.
i
All references in this prospectus to “$”, “US$” and “U.S. dollars” mean U.S. dollars and all references to “A$” mean Australian dollars, unless otherwise noted. Throughout this prospectus, all references to “ADSs” mean American depositary shares, each of which represents of our ordinary shares, no par value, and all references to “ADRs” mean the American depositary receipts that evidence the ADSs.
This prospectus contains translations of some Australian dollar amounts into U.S. dollars. Except as otherwise stated in this prospectus, all translations from Australian dollars to U.S. dollars are based on the exchange rate of US$0.7702 to A$1.00 published by the Reserve Bank of Australia as of December 31, 2020. No representation is made that the Australian dollar amounts referred to in this prospectus could have been or could be converted into U.S. dollars at such rate.
“Incannex”, the Incannex logo and other trademarks or service marks of Incannex appearing in this prospectus are the property of Incannex or its subsidiaries. Solely for convenience, the trademarks, service marks and trade names referred to in this prospectus are listed without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their right thereto. All other trademarks, trade names and service marks appearing in this prospectus are the property of their respective owners.
ii
This summary highlights selected information contained elsewhere in this prospectus. This summary does not contain all of the information you should consider before investing in the ADSs. You should read this entire prospectus, and the registration statement of which this prospectus is a part, including “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and our consolidated financial statements and the related notes included elsewhere in this prospectus, before making an investment decision. Unless otherwise indicated or the context otherwise requires, “Incannex,” the “Company,” “our company,” “we,” “us” and “our” refer to Incannex Healthcare Limited and its consolidated subsidiary, taken as a whole.
Overview
Our legal name is Incannex Healthcare Limited (“Incannex”). We were incorporated in Australia in April 2001 under the name Mount Magnet South Limited. In November 2016, we changed name to Impression Healthcare Limited, and in June 2020, to Incannex Healthcare Limited. Incannex is listed on the ASX under the symbol “IHL”.
Since 2019, we have been conducting research and development for synthetic cannabinoid pharmaceutical products and psychedelic medicine therapies for treatment of a range of indications. Our mission is to create pharmaceutical drugs and therapies for patients that we believe have unmet medical needs. We aim to be recognized as a leading specialty drug development company at the forefront of innovation, committed to restoring health and transforming the lives of patients through the development of novel pharmaceutical products and treatments.
We are developing targeted fixed-dose combinations containing cannabinoids and approved generic drugs, and psychedelic agents, applying proprietary insights in an effort to create long term value for our patients and shareholders. We focus on clinical indications that we believe represent unmet or inadequately addressed medical needs and also represent compelling commercial opportunities. In particular, we are developing three unique pharmaceutical compositions to target five indications: obstructive sleep apnea (“OSA”), traumatic brain injury (“TBI”)/concussion, rheumatoid arthritis (“RA”), inflammatory bowel disease (“IBD”) and inflammatory lung conditions (“ARDS”, “COPD”, Asthma, Bronchitis). We are also developing a treatment for generalized anxiety disorder (“GAD”) utilizing psilocybin combined with innovative psychotherapy methods. Each indication represents a major global market that currently has either no, or limited, existing registered pharmacotherapy (drug) treatments available to the public. To protect our investment in each of these development programs we have been implementing a strong patent filing strategy as we develop our drug candidates in conjunction with our medical and scientific advisory board. The novelty and inventiveness of our cannabinoid products and methods to treat the target indications has been confirmed in international search reports on our filed PCT applications.
We are pursuing FDA registration and marketing approval for each product and therapy under development. As each of our drug candidates is targeting regulatory approval from the FDA, the safety and efficacy must be demonstrated using robust data from in-human clinical trials. Clinical development is an iterative process of clinical trials of increasing size, initially focusing on safety with efficacy becoming an increasing goal as the program progresses. However, our strategy of combining cannabinoids with approved generic drugs allows us to rely on historic, or published data to address some of the key clinical development questions, which in turn permits us to omit or combine clinical studies that would usually be required for approval and registration. We aim to open investigational new drug (IND) applications for each of our development programs by the end of 2022. Clinical trials would then follow with the goal of FDA approval of the applicable drug candidate in the 3-5 years subsequent to the approval of the IND.
Developing drug products containing cannabinoids and psychedelics could result in a more burdensome regulatory process because cannabis, THC, and psychedelics are currently listed as schedule 1 controlled substances by the DEA. This means that they are considered to have no currently accepted medical use and a high potential for abuse. Cannabis, THC and psychedelics have not been legalized for either recreational or medical use in the majority of U.S. states, however, certain states allow the use of cannabis and THC for medical use, and others for medical and recreational use, while Oregon has gone as far as legalizing the recreational use of cannabis, THC and certain psychedelics, including psilocybin. However, FDA approved CBD products containing less than 0.1 % THC where the CBD is extracted from plant material are included in schedule V, which is reserved for drugs with the lowest chance of abuse. CBD manufactured synthetically, that is not extracted from plants, is not currently scheduled. Our CBD drug candidates all use synthetic CBD. Dronabinol, a synthetic form of THC approved by the FDA for treatment of anorexia associated with weight loss in patients with Acquired Immune Deficiency Syndrome (AIDS) as well as
1
nausea and vomiting associated with cancer chemotherapy is included in schedule 3. Our THC containing product uses dronabinol and is expected to be considered a schedule III product. Psilocybin is a schedule I substance but there is substantial peer reviewed literature to support that there is a low chance for abuse. It is expected that once the FDA approves a psilocybin drug product, such as our drug candidate targeting GAD, this will trigger a rescheduling.
Each of our drug candidates is still in the development stage, as we have not initiated an FDA clinical trial as it relates to any drug candidate, nor have we submitted an IND to the FDA for any drug candidate. We must first submit an IND application for each drug candidate before we can initiate a clinical trial with the FDA. In Australia, proof of concept Phase 2 clinical trials are underway for OSA and GAD, Phase 1 pharmacokinetic and safety clinical trials are underway for RA, IBD and inflammatory lung diseases and in depth pre-clinical studies are underway for TBI. To date we have engaged researchers from Monash University, The Alfred Hospital and the University of Western Australia Centre for Sleep Science to conduct our Australian clinical trials. See “Business” section for more information.
Our current cash position is sufficient to complete the studies that have already commenced as well as the pivotal Phase 2 clinical trial for OSA. Future capital raises, including this offer of ADSs will be required to fund additional Phase 1, 2 and 3 studies as well as other development activities required by regulatory bodies, such as the FDA.
To achieve our commercial goals, we intend to advance our novel investigational drug candidates towards approval in the United States and elsewhere. We plan to take advantage of accelerated commercialization pathway options, such as breakthrough designation, accelerated approval, priority review, and/or fast track, to reduce the time and cost of development. However, we have not yet approached the FDA regarding accelerated approval pathways for our products and the FDA has not given any indications that our products will receive these designations. We intend to develop future clinical candidates that target unmet medical needs. We also will continue to maintain a strong intellectual property portfolio to protect our assets in key global markets, including the United States, Europe, Japan, and Israel.
Corporate Information
Our registered office is located at Suite 15, Level 12, 401 Docklands Drive, Docklands 3008, Victoria, Australia and our telephone number is +61 409 840 786. Our website address is www.incannex.com.au. The information on, or accessible through, our website is not part of this prospectus. We have included our website address in this prospectus solely as an inactive textual reference. All information we file with the U.S. Securities and Exchange Commission (“SEC”) is available through the SEC’s Electronic Data Gathering, Analysis and Retrieval system, which may be accessed through the SEC’s website at www.sec.gov.
Implications of Being an Emerging Growth Company
As a company with less than $1.07 billion in revenue during our last fiscal year, we qualify as an “emerging growth company” as defined in the JOBS Act. As an emerging growth company, we may take advantage of specified reduced disclosure and other requirements that are otherwise applicable generally to public companies. These provisions include:
• exemption from the auditor attestation requirement of Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, in the assessment of our internal controls over financial reporting; and
• to the extent that we no longer qualify as a foreign private issuer, (i) reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and (ii) exemptions from the requirements of holding a non-binding advisory vote on executive compensation, including golden parachute compensation.
We may take advantage of these exemptions until such time that we are no longer an emerging growth company. Accordingly, the information that we provide shareholders and holders of the ADSs may be different than you might obtain from other public companies. We will cease to be an emerging growth company upon the earliest to occur of (i) the last day of the fiscal year in which we have more than $1.07 billion in annual revenue; (ii) the last day of the fiscal year in which we qualify as a “large accelerated filer”; (iii) the date on which we have, during the previous three-year period, issued more than $1.0 billion in non-convertible debt securities; and (iv) the last day of the fiscal year in which the fifth anniversary of this offering occurs.
2
Implications of Being a Foreign Private Issuer
We are also considered a “foreign private issuer” under U.S. securities laws. In our capacity as a foreign private issuer, we are exempt from certain rules under the Exchange Act that impose certain disclosure obligations and procedural requirements for proxy solicitations under Section 14 of the Exchange Act. In addition, our senior management, the members of our board of directors and our principal shareholders are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and the rules under the Exchange Act with respect to their purchases and sales of our securities. Moreover, we are not required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. companies whose securities are registered under the Exchange Act. In addition, we are not required to comply with Regulation FD, which restricts the selective disclosure of material information.
We may take advantage of these exemptions until such time as we are no longer a foreign private issuer. We will remain a foreign private issuer until such time that 50% or more of our outstanding voting securities are held by U.S. residents and any of the following three circumstances applies: (i) the majority of the members of board of directors or our senior management are U.S. citizens or residents; (ii) more than 50% of our assets are located in the United States; or (iii) our business is administered principally in the United States.
Risk Factors Summary
Our business is subject to a number of risks of which you should be aware prior to making a decision to invest in our ADSs. You should carefully consider all of the information set forth in this prospectus and, in particular, should evaluate the specific factors set forth in the section titled “Risk Factors” before deciding whether to invest in our ADSs. Among these important risks are, but not limited to, the following:
• We have a history of operating losses and may not achieve or maintain profitability in the future.
• We currently have no source of product revenue and may never become profitable.
• We will require additional financing and may be unable to raise sufficient capital, which could have a material impact on our research and development programs or commercialization of our drug candidates.
• We may find it difficult to enroll patients in our current and any future clinical trials, and patients could discontinue their participation in our current and any future clinical trials, which could delay or prevent our current and any future clinical trials of our drug candidates and make those trials more expensive to undertake.
• Positive results from preclinical studies of our drug candidates are not necessarily predictive of the results of our planned clinical trials of our drug candidates.
• Ongoing and future clinical trials of drug candidates may not show sufficient safety and efficacy to obtain requisite regulatory approvals for commercial sale.
Even if our drug candidates receive regulatory approval, they may still face development and regulatory difficulties that may delay or impair future sales of drug candidates.
• Because we rely on third party manufacturing and supply partners, our supply of research and development, preclinical and clinical development materials may become limited or interrupted or may not be of satisfactory quantity or quality.
• Future potential sales of our drug candidates may suffer if they are not accepted in the marketplace by physicians, patients and the medical community.
• Our drug candidates will be subject to controlled substance laws and regulations. Failure to receive necessary approvals may delay the launch of our drug candidates and failure to comply with these laws and regulations may adversely affect the results of our business operations.
3
• Intellectual property rights of third parties could adversely affect our ability to commercialize our drug candidates, such that we could be required to litigate with or obtain licenses from third parties in order to develop or market our drug candidates. Such litigation or licenses could be costly or not available on commercially reasonable terms.
• Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
• The trading price of the ADSs may be volatile, and purchasers of the ADSs could incur substantial losses.
• There has been no prior market for the ADSs and an active and liquid market for our securities may fail to develop, which could harm the market price of the ADSs.
• You will experience immediate and substantial dilution in the net tangible book value of the ADSs you purchase in this offering.
• As long as we remain subject to the rules of the ASX and Nasdaq, we will be unable to access equity capital without shareholder approval if such equity capital sales would result in an equity issuance above regulatory thresholds and, consequently, we may be unable to obtain financing sufficient to sustain our business if we are unsuccessful in soliciting requisite shareholder approvals.
• Our ADS holders are not shareholders and do not have shareholder rights.
• Australian takeovers laws may discourage takeover offers being made for us or may discourage the acquisition of large numbers of our shares.
• U.S. investors may have difficulty enforcing civil liabilities against our company, our directors or member of senior management and the experts named in this prospectus.
Special Note Regarding Forward-Looking Statements
This prospectus contains forward-looking statements about us and our industry that involve substantial risks and uncertainties. All statements other than statements of historical facts contained in this prospectus, including statements regarding our future results of operations, financial condition, business strategy and plans and objectives of management for future operations, are forward-looking statements. In some cases, you can identify forward-looking statements because they contain words such as “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” or “would,” or the negative of these words or other similar terms or expressions.
We have based these forward-looking statements largely on our current expectations and projections about future events and trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. These forward-looking statements are subject to a number of known and unknown risks, uncertainties, other factors and assumptions, including the risks described in “Risk Factors” and elsewhere in this prospectus, regarding, among other things:
• our product development and business strategy, including the potential size of the markets for our drug candidates and future development and/or expansion of our drug candidates in our markets;
• our current and future research and development activities, including clinical testing and manufacturing and the costs and timing thereof;
• the impact that the COVID-19 pandemic could have on business operations;
• sufficiency of our cash resources;
• our ability to commercialize drug candidates and generate product revenues;
• our ability to raise additional funding when needed;
4
• any statements concerning anticipated regulatory activities or licensing or collaborative arrangements, including our ability to obtain regulatory clearances;
• our research and development and other expenses;
• our operations and intellectual property risks;
• our ability to remain compliant with the Australian Securities Exchange (“ASX”) and Nasdaq’s continuing listing standards;
• any statement of assumptions underlying any of the foregoing; and
• other risks and uncertainties, including those listed under “Risk Factors.”
These risks are not exhaustive. Other sections of this prospectus may include additional factors that could harm our business and financial performance. New risk factors may emerge from time to time and it is not possible for our management to predict all risk factors, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in, or implied by, any forward-looking statements.
You should not rely on forward-looking statements as predictions of future events. We have based the forward-looking statements contained in this prospectus primarily on our current expectations and projections about future events and trends that we believe may affect our business, financial condition and operating results. We undertake no obligation to update any forward-looking statements made in this prospectus to reflect events or circumstances after the date of this prospectus or to reflect new information or the occurrence of unanticipated events, except as required by law. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based on information available to us as of the date of this prospectus. While we believe that information provides a reasonable basis for these statements, that information may be limited or incomplete. Our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all relevant information. These statements are inherently uncertain, and investors are cautioned not to unduly rely on these statements.
You should read this prospectus and the documents that we reference in this prospectus and have filed as exhibits to the registration statement of which this prospectus is a part with the understanding that our actual future results, levels of activity, performance and achievements may be different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
5
|
ADSs offered by us |
ADSs. |
|
|
Option to purchase additional ADSs |
The underwriter has an option for a period of 30 days from the date of this prospectus to purchase up to additional ADSs. |
|
|
Ordinary shares to be outstanding after this offering, including shares underlying ADSs |
shares (or shares if the underwriter exercises its option to purchase additional ADSs in full). |
|
|
American depositary shares |
Each ADS represents 50 ordinary shares. The ADSs are issued by the depositary. You will have the rights of an ADS holder as provided in the deposit agreement among us, the depositary and all owners and holders of ADSs issued thereunder. The depositary, through its custodian, will be the holder of the ordinary shares underlying the ADSs. |
|
|
You may surrender your ADSs to the depositary for cancellation to receive the ordinary shares underlying your ADSs. The depositary will charge you a fee for such a cancellation. |
||
|
We may amend or terminate the deposit agreement for any reason without your consent. Any amendment that imposes or increases fees or charges or that materially prejudices any substantial existing right you have as an ADS holder will not become effective as to outstanding ADSs until 30 days after notice of the amendment is given to ADS holders. If an amendment becomes effective, you will be bound by the deposit agreement as amended if you continue to hold your ADSs. |
||
|
To better understand the terms of the ADSs, you should carefully read the section titled “Description of American Depositary Shares.” We also encourage you to read the deposit agreement, which is filed as an exhibit to the registration statement of which this prospectus forms a part. |
||
|
Depositary |
Deutsche Bank Trust Company Americas. |
|
|
Use of proceeds |
We estimate that the net proceeds from the sale of the ADSs that we are selling in this offering will be approximately US$ million (or approximately US$ million if the underwriter’s option to purchase additional ADSs is exercised in full), based upon an assumed initial public offering price of $ per ADS, after giving effect to the Australian dollar/U.S. dollar exchange rate of as of , 2021, and an ADS-to-ordinary share ratio of 1-to-50, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. |
|
|
We intend to use the net proceeds from this offering, together with our existing cash, to further our clinical trials, for working capital and other general corporate purposes. See “Use of Proceeds” for additional information. |
6
|
Underwriter Warrants |
Upon the closing of this offering, we will issue warrants to the underwriter (the “Underwriter Warrants”) entitling it to purchase a number of ordinary shares, represented by ADSs, equal to 7.5% of the ADSs sold in this offering by us, in three tranches of 2.5% each: (i) the first tranche representing 2.5% of the ADSs sold in this offering having an exercise price equal to 120% of the public offering price of the ADSs in this offering, (ii) the second tranche representing 2.5% of the ADSs sold in this offering having an exercise price equal to 135% of the public offering price of the ADSs in this offering and (iii) the third tranche representing 2.5% of the ADSs sold in this offering having an exercise price equal to 150% of the public offering price of the ADSs in this offering. All ordinary shares will be in the form of ADSs, with each ADS representing 50 ordinary shares. The Underwriter Warrants will expire three (3) years after the effective date of the registration statement of which this prospectus forms a part. See “Underwriting.” |
|
|
Risk factors |
See “Risk Factors” and the other information included in this prospectus for a discussion of the risks you should carefully consider before investing in the ADSs. |
|
|
Proposed Nasdaq Capital Market symbol for the ADSs |
“IXHL” |
|
|
Australian Stock Exchange symbol for our ordinary shares |
“IHL” |
The number of ordinary shares (including ordinary shares underlying ADSs) that will be outstanding after this offering is based on 1,068,411,224 ordinary shares outstanding as of June 30, 2021 and excludes 326,437,328 ordinary shares issuable upon the exercise of outstanding options as of June 30, 2021, with a weighted-average exercise price of A$0.1655 per ordinary share.
In addition, unless we specifically state otherwise, the information in this prospectus assumes (i) no exercise by the underwriter of (a) its option to purchase up to additional ADSs or (b) their warrants to purchase (x) ADSs at an exercise price equal to 120% of the initial public offering price per ADS, (y) ADSs at an exercise price equal to 135% of the initial public offering price per ADS and (z) ADSs at an exercise price equal to 150% of the initial public offering price per ADS and (ii) no exercise of outstanding options to purchase ordinary shares.
7
Summary Consolidated Financial Data
The following tables summarize our consolidated financial and other data. The summary consolidated statement of profit or loss and other comprehensive income data for the six months ended December 31, 2020 and 2019 and the consolidated statement of financial position data as of December 31, 2020 have been derived from our unaudited consolidated financial statements included elsewhere in this prospectus. The summary consolidated statement of profit or loss and other comprehensive income data for the years ended June 30, 2020 and 2019 have been derived from our audited consolidated financial statements included elsewhere in this prospectus. Our audited consolidated financial statements have been prepared in accordance with IFRS, as issued by the IASB, as of and for the years ended June 30, 2020 and 2019.
You should read the consolidated financial and other data set forth below in conjunction with our consolidated financial statements and the accompanying notes, the information in “Selected Consolidated Financial and Other Data” and the information in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” contained elsewhere in this prospectus.
Consolidated Statement of Profit or Loss and Other Comprehensive Income Data
|
Six months ended December 31, |
Year ended June 30, |
|||||||||||
|
2020 |
2019 |
2020 |
2019 |
|||||||||
|
(in A$, except share amounts) |
(in A$, except share amounts) |
|||||||||||
|
Revenue |
1,177,163 |
|
7,350 |
|
604,884 |
|
— |
|
||||
|
Product costs |
(537,939 |
) |
(8,450 |
) |
(450,345 |
) |
— |
|
||||
|
Research and development costs |
(2,039,147 |
) |
(313,426 |
) |
(2,110,639 |
) |
(736,140 |
) |
||||
|
Loss after income tax expense from continuing operations |
(2,889,389 |
) |
(1,925,473 |
) |
(3,929,284 |
) |
(1,426,198 |
) |
||||
|
Net loss |
(2,889,389 |
) |
(2,212,004 |
) |
(4,697,636 |
) |
(2,718,399 |
) |
||||
|
Loss per share from continuing operations – basic and diluted (in A$ cents) |
(0.32 |
) |
(0.30 |
) |
(0.57 |
) |
(0.32 |
) |
||||
|
Loss per share from continuing operations and discontinued operations – basic and diluted (in A$ cents) |
(0.32 |
) |
(0.34 |
) |
(0.69 |
) |
(0.61 |
) |
||||
|
Weighted average number of ordinary shares outstanding – basic and diluted |
902,054,732 |
|
649,048,889 |
|
684,035,399 |
|
447,439,263 |
|
||||
|
Dividends per share |
— |
|
— |
|
— |
|
— |
|
||||
8
Consolidated Statement of Financial Position Data(1)(2)
|
As of December 31, 2020 |
||||||||||
|
Actual |
As Adjusted(1)(2) |
|||||||||
|
(in A$) |
(in US$) |
(in A$) |
in US$) |
|||||||
|
Cash |
11,840,308 |
|
9,119,405 |
|
||||||
|
Net assets |
11,802,503 |
|
9,090,287 |
|
||||||
|
Total assets |
12,180,952 |
|
9,381,769 |
|
||||||
|
Total liabilities |
378,449 |
|
291,481 |
|
||||||
|
Accumulated losses |
(35,407,592 |
) |
(27,270,927 |
) |
||||||
|
Issued capital |
45,076,484 |
|
34,717,907 |
|
||||||
____________
(1) The as adjusted statement of financial position data give effect to our receipt of net proceeds from the issuance and sale of ADSs at the assumed initial offering price of US$ per ADS, after giving effect to the ADS-to-ordinary share ratio of 1-to-50, after deducting underwriting commissions and estimated offering expenses payable by us.
(2) Each $1.00 increase or decrease in the assumed initial public offering price of US$ per ADS, after giving effect to the ADS-to-ordinary share ratio of 1-to-50, would increase or decrease, respectively, the amount of cash, working capital, total assets and total equity by A$ million (or US$ million), assuming the number of ADSs offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. We may also increase or decrease the number of ADSs we are offering. An increase or decrease of 1,000,000 in the number of ADSs we are offering would increase or decrease the amount of cash, working capital, total assets and total equity by A$ million (or US$ million), assuming the assumed initial public offering price per ADS remains the same and after deducting underwriting discounts and commissions. The as adjusted information is illustrative only and will be adjusted based on the actual initial public offering price and other terms of this offering determined at pricing.
9
Investing in the ADSs involves a high degree of risk. You should consider and read carefully all of the risks and uncertainties described below, as well as other information included in this prospectus, including our consolidated financial statements and related notes included elsewhere in this prospectus, before making an investment decision. If any of the following risks actually occur, it could harm our business, prospects, results of operations and financial condition. In such event, the trading price of the ADSs could decline and you might lose all or part of your investment.
Risks Related to Our Business
We have a history of operating losses and may not achieve or maintain profitability in the future.
We have experienced significant recurring operating losses and negative cash flows from operating activities since inception. For example, for the years ended June 30, 2020 and 2019, we had net losses of approximately A$4.7 million and approximately A$2.7 million, respectively, and approximately A$2.9 million and approximately A$2.2 million for the six months ended December 31, 2020 and 2019, respectively. As of December 31, 2020, we had accumulated losses of approximately A$35.4 million.
We are a clinical stage pharmaceutical development company and the success of our drug candidates is therefore uncertain. We focus on medicinal synthetic cannabinidiol pharmaceutical products and psychedelic medicine therapies.
We expect to continue to incur losses from operations for the foreseeable future and expect the costs of drug development to increase in the future as more patients are recruited to the clinical trials. In particular, we expect to continue to incur significant losses in the development of our clinical trials and drug candidates. Because of the numerous risks and uncertainties associated with the development, manufacturing, sales and marketing of our drug candidates, we may experience larger than expected future losses and may never become profitable.
Moreover, there is a substantial risk that we, or our development partners, may not be able to complete the development of our current drug candidates or develop other pharmaceutical products. It is possible that none of them will be successfully commercialized, which would prevent us from ever achieving profitability.
The increase in expenses may adversely impact our business if our sources of funding and revenue are insufficient.
We anticipate that as the costs related to the development of our clinical trials will increase, we will require additional funds to achieve our long-term goals of commercialization and further development of our drug candidates. In addition, we will require funds to pursue regulatory applications, defend intellectual property rights, contract manufacturing capacity, potentially develop marketing and sales capability and fund operating expenses. We intend to seek such additional funding through public or private financings and/or through licensing of our assets or other arrangements with corporate partners. However, such financing, licensing opportunities or other arrangements may not be available from any sources on acceptable terms, or at all. Any shortfall in funding could result in us having to curtail or cease our research and development activities, thereby harming our business, financial condition and results of operations.
In addition, because of the numerous risks and uncertainties associated with the development of our drug candidates, we are unable to predict the timing or amount of increased expenses, or when, or if, we will be able to achieve or maintain profitability. Our expenses could significantly increase beyond current expectations if the applicable regulatory authorities require further studies in addition to those currently anticipated. In any case, even if our drug candidates are approved for commercial sale, we anticipate incurring significant costs associated with the commercial launch of such drug candidates and there can be no guarantee that we will ever generate significant revenues.
We currently have no source of product revenue and may never become profitable.
Our drug candidates have not been approved for commercial sale, and we expect it to be several years before they are approved, if ever, and we are able to commence sales of our drug candidates. To date, we have not generated any revenue from the licensing or commercialization of our drug candidates and do not expect to receive revenue
10
from them for a number of years, if ever. We will not be able to generate product revenue unless and until our current drug candidates or any future drug candidates, alone or with future partners, successfully completes clinical trials, receives regulatory approval and is successfully commercialized. Although we may seek to obtain revenue from collaboration or licensing agreements with third parties, we currently have no such agreements that could provide us with material, ongoing future revenue and we may never enter into any such agreements.
We will require additional financing and may be unable to raise sufficient capital, which could have a material impact on our research and development programs or commercialization of our drug candidates.
We have historically devoted most of our financial resources to research and development, including pre-clinical and clinical development activities. To date, we have financed a significant amount of our operations through equity financings. The amount of our future net losses will depend, in part, on the rate of our future expenditures and our ability to obtain funding through equity or debt financings or strategic collaborations. The amount of such future net losses, as well as the possibility of future profitability, will also depend on our success in developing and commercializing products that generate significant revenue. Our failure to become and remain profitable would depress the value of our ADSs and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings or even continue our operations.
We anticipate that our expenses will increase substantially for the foreseeable future if, and as, we:
• continue our research and preclinical and clinical development of our drug candidates;
• expand the scope of our current proposed clinical studies for our drug candidates;
• initiate additional preclinical, clinical or other studies for our drug candidates;
• change or add additional manufacturers or suppliers;
• seek regulatory and marketing approvals for our drug candidates that successfully complete clinical studies;
• seek to identify and validate additional drug candidates;
• acquire or in-license other drug candidates and technologies;
• maintain, protect and expand our intellectual property portfolio;
• attract and retain skilled personnel;
• create additional infrastructure to support our operations as a publicly quoted company and our product development and planned future commercialization efforts;
• add an internal sales force; and
• experience any delays or encounter issues with any of the above.
Until our drug candidates become commercially available, we will need to obtain additional funding in connection with the further development of our drug candidates. Our ability to obtain additional financing will be subject to a number of factors, including market conditions, our operating performance and investor sentiment. As such, additional financing may not be available to us when needed, on acceptable terms, or at all. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts or obtain funds by entering agreements on unattractive terms.
Furthermore, any additional equity fundraising in the capital markets may be dilutive for shareholders and any debt-based funding may bind us to restrictive covenants and curb our operating activities and ability to pay potential future dividends even when profitable. We cannot guarantee that future financing will be available in sufficient amounts or on acceptable terms, if at all. If we are unable to raise additional capital in sufficient amounts or on acceptable terms, we will be prevented from pursuing research and development efforts. This could harm our business, operating results and financial condition and cause the price of our ADSs to fall.
11
If we are unable to secure sufficient capital to fund our operations, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to third parties to develop and market drug candidates that we would otherwise prefer to develop and market ourselves. For example, additional strategic collaborations could require us to share commercial rights to our drug candidates with third parties in ways that we do not intend currently or on terms that may not be favorable to us. Moreover, we may also have to relinquish valuable rights to our technologies, future revenue streams, research programs or drug candidates or grant licenses on terms that may not be favorable to us.
We may find it difficult to enroll patients in our current and any future clinical trials, and patients could discontinue their participation in our current and any future clinical trials, which could delay or prevent our current and any future clinical trials of our drug candidates and make those trials more expensive to undertake.
Identifying and qualifying patients to participate in current and any future clinical trials of our drug candidates is critical to our success. The timing of our clinical trials depends on the speed at which we can recruit patients to participate in testing our drug candidates. Patients may be unwilling to participate in any future clinical trials because of negative publicity from adverse events in the biotechnology industry. Patients may be unavailable for other reasons, including competitive clinical trials for similar patient populations, and the timeline for recruiting patients, conducting trials and obtaining regulatory approval of potential products may be delayed. If we have difficulty enrolling a sufficient number of patients to conduct any future clinical trials as planned, we may need to delay, limit or discontinue those clinical trials. Clinical trial delays could result in increased costs, slower product development, setbacks in testing the safety and effectiveness of our technology or discontinuation of the clinical trials altogether.
Any failure to implement our business strategy could negatively impact our business, financial condition and results of operations.
The development and commercialization of our drug candidates is subject to many risks, including:
• additional clinical or pre-clinical trials may be required beyond what we currently expect;
• regulatory authorities may disagree with our interpretation of data from our preclinical studies and clinical studies or may require that we conduct additional studies;
• regulatory authorities may disagree with our proposed design of future clinical trials;
• regulatory authorities may delay approval of our drug candidates, thus preventing milestone payments from our collaboration partners;
• regulatory authorities may not accept data generated at our clinical study sites;
• we may be unable to obtain and maintain regulatory approval of our drug candidate in any jurisdiction;
• the prevalence and severity of any side effects of any drug candidate could delay or prevent commercialization, limit the indications for any approved drug candidate, require the establishment of a risk evaluation and mitigation strategy, or cause an approved drug candidate to be taken off the market;
• regulatory authorities may identify deficiencies in manufacturing processes;
• regulatory authorities may change their approval policies or adopt new regulations;
• the third party manufacturers we expect to depend on to supply or manufacture our drug candidates may not produce adequate supply;
• we, or our third party manufacturers, may not be able to source or produce current Good Manufacturing Practice (cGMP) materials for the production of our drug candidates;
• we may not be able to manufacture our drug candidates at a cost or in quantities necessary to make commercially successful products;
• we may not be able to obtain adequate supply of our drug candidates for our clinical trials;
12
• we may experience delays in the commencement of, enrolment of patients in and timing of our clinical trials;
• we may not be able to demonstrate that our drug candidates are safe and effective as a treatment for its indications to the satisfaction of regulatory authorities, and we may not be able to achieve and maintain compliance with all regulatory requirements applicable to our drug candidates;
• we may not be able to maintain a continued acceptable safety profile of our products following approval;
• we may be unable to establish or maintain collaborations, licensing or other arrangements;
• the market may not accept our drug candidates;
• we may be unable to establish and maintain an effective sales and marketing infrastructure, either through the creation of a commercial infrastructure or through strategic collaborations, and the effectiveness of our own or any future strategic collaborators’ marketing, sales and distribution strategy and operations will affect our profitability;
• we may experience competition from existing products or new products that may emerge;
• we and our licensors may be unable to successfully obtain, maintain, defend and enforce intellectual property rights important to protect our drug candidates; and
• we may not be able to obtain and maintain coverage and adequate reimbursement from third party payors.
If any of these risks materializes, we could experience significant delays or an inability to successfully develop and commercialize our drug candidates we or our partners may develop, which would have a material adverse effect on our business, financial condition and results of operations.
Positive results from preclinical studies of our drug candidates are not necessarily predictive of the results of our planned clinical trials of our drug candidates.
Positive results in preclinical proof of concept and animal studies of our drug candidates may not result in positive results in clinical trials in humans. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical trials after achieving positive results in preclinical development or early stage clinical trials, and we cannot be certain that we will not face similar setbacks. These setbacks can be caused by, among other things, preclinical findings made while clinical trials were underway or safety or efficacy observations made in clinical trials, including adverse events. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their drug candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to obtain FDA or other regulatory authority approval. If we fail to produce positive results in our clinical trials of our drug candidates, the development timeline and regulatory approval and commercialization prospects for our drug candidates, and, correspondingly, our business and financial prospects, would be negatively impacted.
Ongoing and future clinical trials of drug candidates may not show sufficient safety and efficacy to obtain requisite regulatory approvals for commercial sale.
Phase I and Phase II clinical trials are not primarily designed to test the efficacy of a drug candidate but rather to test safety and to understand the drug candidate’s side effects at various doses and schedules. Furthermore, success in preclinical and early clinical trials does not ensure that later large-scale trials will be successful nor does it predict final results. Acceptable results in early trials may not be repeated in later trials. Further, Phase III clinical trials may not show sufficient safety or efficacy to obtain regulatory approval for marketing. In addition, clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. Negative or inconclusive results or adverse medical events during a clinical trial could require that the clinical trial be redone or terminated. The length of time necessary to complete clinical trials and to submit an application for marketing approval by applicable regulatory authorities may also vary significantly based on the type, complexity and novelty of the drug candidate involved, as well as other factors. If we suffer any significant delays, quality issues, setbacks or negative results in, or termination of, our clinical trials, we may be unable to continue the development of our drug candidates or generate revenue and our business may be severely harmed.
13
If we do not obtain the necessary regulatory approvals, we will be unable to commercialize our drug candidates.
The clinical development, manufacturing, sales and marketing of our drug candidates are subject to extensive regulation by regulatory authorities in the United States, the United Kingdom, the European Union, Australia and elsewhere. Despite the substantial time and expense invested in preparation and submission of a Biologic License Application or equivalents in other jurisdictions, regulatory approval is never guaranteed. The number, size and design of preclinical studies and clinical trials that will be required will vary depending on the product, the disease or condition for which the product is intended to be used and the regulations and guidance documents applicable to any particular product. Additionally, during the review process and prior to approval, the FDA and/or other regulatory bodies may require additional data, including with respect to whether our products have abuse potential, which may delay approval and any potential controlled substance scheduling processes. The FDA or other regulators can delay, limit or deny approval of a product for many reasons, including, but not limited to, the fact that regulators may not approve our or a third party manufacturer’s processes or facilities or that new laws may be enacted or regulators may change their approval policies or adopt new regulations requiring new or different evidence of safety and efficacy for the intended use of a product.
Successful results in clinical trials and in the subsequent application for marketing approval are not guaranteed. If we are unable to obtain regulatory approvals, we will not be able to generate revenue from our drug candidates. Even if we receive regulatory approval for any of our drug candidates, our profitability will depend on our ability to generate revenues from their sale or the licensing of our technology.
Even if our drug candidates receive regulatory approval, it may still face development and regulatory difficulties that may delay or impair future sales of drug candidates.
Even if we or our licensing partners receive regulatory approval to sell any drug candidates, the relevant regulatory authorities may, nevertheless, impose significant restrictions on the indicated uses, manufacturing, labelling, packaging, adverse event reporting, storage, advertising, promotion and record keeping or impose ongoing requirements for post-approval studies. In addition, regulatory agencies subject a marketed product, its manufacturer and the manufacturer’s facilities to continual review and periodic inspections. Previously unknown problems with the drug candidate, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the product, and could include withdrawal of the product from the market. In addition, new statutory requirements may be enacted or additional regulations may be enacted that could prevent or delay regulatory approval of our drug candidates.
We have limited manufacturing experience with our drug candidates.
We have no manufacturing capabilities and are dependent on third parties for cost effective manufacture and manufacturing process development of the company’s drug candidates. Problems with third party manufacturers or the manufacturing process, or the scaling up of manufacturing activities as such may delay clinical trials and commercialization of our drug candidates.
To the extent we rely significantly on contractors, we will be exposed to risks related to the business and operational conditions of our contractors.
We are a small company, with few internal staff and limited facilities. We are and will be required to rely on a variety of contractors to manufacture and transport our drug candidates, to perform clinical testing and to prepare regulatory dossiers. Adverse events that affect one or more of our contractors could adversely affect us, such as:
• a contractor is unable to retain key staff that have been working on our drug candidates;
• a contractor is unable to sustain operations due to financial or other business issues;
• a contractor loses their permits or licenses that may be required to manufacture our drug candidates; or
• errors, negligence or misconduct that occur within a contractor may adversely affect our business.
14
We depend on, and will continue to depend on, collaboration and strategic alliances with third partners. To the extent we are able to enter into collaborative arrangements or strategic alliances, we will be exposed to risks related to those collaborations and alliances.
An important element of our strategy for developing, manufacturing and commercializing our drug candidates is entering into partnerships and strategic alliances with other pharmaceutical companies or other industry participants.
Any partnerships or alliance we have or may have in the future may be terminated for reasons beyond our control or we may not be able to negotiate future alliances on acceptable terms, if at all. These arrangements may result in us receiving less revenue than if we sold our products directly, may place the development, sales and marketing of our products outside of our control, may require us to relinquish important rights or may otherwise be on unfavorable terms. Collaborative arrangements or strategic alliances will also subject us to a number of risks, including the risk that:
• we may not be able to control the amount and timing of resources that our strategic partner/collaborators may devote to the drug candidates;
• strategic partner/collaborators may experience financial difficulties;
• the failure to successfully collaborate with third parties may delay, prevent or otherwise impair the development or commercialization of our drug candidates or revenue expectations;
• products being developed by partners/collaborators may never reach commercial stage resulting in reduced or even no milestone or royalty payments;
• business combinations or significant changes in a collaborator’s business strategy may also adversely affect a collaborator’s willingness or ability to complete their obligations under any arrangement;
• a collaborator could independently move forward with a competing product developed either independently or in collaboration with others, including our competitors; and
• collaborative arrangements are often terminated or allowed to expire, which would delay the development and may increase the cost of developing drug candidates.
Because we rely on third party manufacturing and supply partners, our supply of research and development, preclinical and clinical development materials may become limited or interrupted or may not be of satisfactory quantity or quality.
We rely on third party supply and manufacturing partners to manufacture and supply the materials for our research and development and preclinical and clinical study supplies. We do not own manufacturing facilities or supply sources for such materials.
There can be no assurance that our supply of research and development, preclinical and clinical development biologics and other materials will not be limited, interrupted or restricted in certain geographic regions, be of satisfactory quality or continue to be available at acceptable prices. Replacement of a third party manufacturer could require significant effort, cost and expertise because there may be a limited number of qualified replacements.
The manufacturing process for a drug candidate is subject to FDA and foreign regulatory authority review. Suppliers and manufacturers must meet applicable manufacturing requirements and undergo rigorous facility and process validation tests required by regulatory authorities in order to comply with regulatory standards. In the event that any of our suppliers or manufacturers fails to comply with such requirements or to perform its obligations to us in relation to quality, timing or otherwise, or if our supply of components or other materials becomes limited or interrupted for other reasons, we may be forced to manufacture the materials ourselves or enter into an agreement with another third party, which would be costly and delay any future clinical trials.
Further, if any third-party provider fails to meet its obligations to manufacture our products, or fails to maintain or achieve satisfactory regulatory compliance, the development of such substances and the commercialization of any therapies, if approved, could be stopped, delayed or made commercially unviable, less profitable or may result in enforcement actions against us.
15
Our research and development efforts will be jeopardized if we are unable to retain key personnel and cultivate key academic and scientific collaborations.
Changes in our senior management may be disruptive to our business and may adversely affect our operations. For example, when we have changes in senior management positions, we may elect to adopt different business strategies or plans. Any new strategies or plans, if adopted, may not be successful and if any new strategies or plans do not produce the desired results, our business may suffer.
Moreover, competition among biotechnology and pharmaceutical companies for qualified employees is intense and as such we may not be able to attract and retain personnel critical to our success. Our success depends on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel, manufacturing personnel, sales and marketing personnel and on our ability to develop and maintain important relationships with clinicians, scientists and leading academic and health institutions. If we fail to identify, attract, retain and motivate these highly skilled personnel, we may be unable to continue our product development and commercialization activities.
In addition, biotechnology and pharmaceutical industries are subject to rapid and significant technological change. Our drug candidates may be or become uncompetitive. To remain competitive, we must employ and retain suitably qualified staff that are continuously educated to keep pace with changing technology, but may not be in a position to do so.
We may encounter difficulties in managing our growth, which could negatively impact our operations.
As we advance our clinical development programs for drug candidates, seek regulatory approval in the United States and elsewhere and increase the number of ongoing product development programs, we anticipate that we will need to increase our product development, scientific and administrative headcount. We will also need to establish commercial capabilities in order to commercialize any drug candidates that may be approved. Such an evolution may impact our strategic focus and our deployment and allocation of resources.
Our ability to manage our operations and growth effectively depends upon the continual improvement of our procedures, reporting systems and operational, financial and management controls. We may not be able to implement administrative and operational improvements in an efficient or timely manner and may discover deficiencies in existing systems and controls. If we do not meet these challenges, we may be unable to execute our business strategies and may be forced to expend more resources than anticipated addressing these issues.
We may acquire additional technology and complementary businesses in the future. Acquisitions involve many risks, any of which could materially harm our business, including the diversion of management’s attention from core business concerns, failure to effectively exploit acquired technologies, failure to successfully integrate the acquired business or realize expected synergies or the loss of key employees from either our business or the acquired businesses.
In addition, in order to continue to meet our obligations as a public listed company in both Australia and the United States and to support our anticipated long-term growth, we will need to increase our general and administrative capabilities. Our management, personnel and systems may not be adequate to support this future growth.
If we are unable to successfully manage our growth and the increased complexity of our operations, our business, financial position, results of operations and prospects may be harmed.
Future potential sales of our drug candidates may suffer if they are not accepted in the marketplace by physicians, patients and the medical community.
There is a risk that our drug candidates may not gain market acceptance among physicians, patients and the medical community, even if they are approved by the regulatory authorities. The degree of market acceptance of any of our approved drug candidates will depend on a variety of factors, including:
• timing of market introduction, number and clinical profile of competitive products;
• our ability to provide acceptable evidence of safety and efficacy and our ability to secure the support of key clinicians and physicians for our drug candidates;
16
• cost-effectiveness compared to existing and new treatments;
• availability of coverage, reimbursement and adequate payment from health maintenance organizations and other third party payers;
• prevalence and severity of adverse side effects; and
• other advantages over other treatment methods.
As controlled substances, the products may generate public controversy. Physicians, patients, payers or the medical community may be unwilling to accept, use or recommend our drug candidates which would adversely affect our potential revenues and future profitability. Adverse publicity or public perception regarding cannabis and psilocybin to our current or future investigational therapies using these substances may negatively influence the success of these therapies.
We face competition from entities that may develop drug candidates for our target disease indications, including companies developing novel treatments and technology platforms based on modalities and technology similar to ours.
The development and commercialization of drug candidates is highly competitive. Multinational pharmaceutical companies and specialized biotechnology companies could develop drug candidates and processes competitive with our drug candidates. Competitive therapeutic treatments include those that have already been approved and accepted by the medical community, patients and third party payers, and any new treatments that enter the market.
There may be a significant number of products that are currently under development, and may become commercially available in the future, for the treatment of conditions for which we are developing, and may in the future try to develop, drug candidates.
Multinational pharmaceutical companies and specialized biotechnology companies could have significantly greater financial, technical, manufacturing, marketing, sales and supply resources and experience than we have. If we successfully obtain approval for any drug candidate, we could face competition based on many different factors, including the safety and effectiveness of our drug candidates, the ease with which our drug candidates can be administered and the extent to which patients accept relatively new routes of administration, the timing and scope of regulatory approvals for these drug candidates, the availability and cost of manufacturing, marketing and sales capabilities, price, reimbursement coverage and patent position.
Competing products could present superior treatment alternatives, including by being more effective, safer, less expensive or marketed and sold more effectively than any products we may develop. Competitive products may make any products we develop obsolete or non-competitive before we recover the expense of developing and commercializing our drug candidates. Such competitors could also recruit our employees, which could negatively impact our level of expertise and our ability to execute our business plan.
If healthcare insurers and other organizations do not pay for our drug candidates or impose limits on reimbursement, our future business may suffer.
Our drug candidates may be rejected by the market due to many factors, including cost. The continuing efforts of governments, insurance companies and other payers of healthcare costs to contain or reduce healthcare costs may affect our future revenues and profitability. In Australia and certain foreign markets, the pricing of pharmaceutical products is subject to government control. We expect initiatives for similar government control to continue in the United States and elsewhere. The adoption of any such legislative or regulatory proposals could harm our business and prospects.
Successful commercialization of our drug candidates will depend in part on the extent to which reimbursement for the cost of our products and related treatment will be available from government health administration authorities, private health insurers and other organizations. Our drug candidates may not be considered cost-effective and reimbursement may not be available to consumers or may not be sufficient to allow our products to be marketed on a competitive basis. Third-party payers are increasingly challenging the price of medical products and treatment.
17
If third party coverage is not available for our drug candidates the market acceptance of these drug candidates will be reduced. Cost-control initiatives could decrease the price we might establish for drug candidates, which could result in product revenues lower than anticipated. If the price for our drug candidates decreases or if governmental and other third-party payers do not provide adequate coverage and reimbursement levels our potential revenue and prospects for profitability will suffer.
We may be exposed to product liability claims which could harm our business.
The testing, marketing and sale of therapeutic products entails an inherent risk of product liability. We rely on a number of third-party researchers and contractors to produce, collect, and analyze data regarding the safety and efficacy of our drug candidates. We also have quality control and quality assurance in place to mitigate these risks, as well as professional liability and clinical trial insurance to cover financial damages in the event that human testing is done incorrectly or the data is analyzed incorrectly.
Notwithstanding our control procedures, we may face product liability exposure related to the testing of our drug candidates in human clinical trials. If any of our drug candidates are approved for sale, we may face exposure to claims by an even greater number of persons than were involved in the clinical trials once marketing, distribution and sales of our drug candidates begin. Regardless of merit or eventual outcome, liability claims may result in:
• decreased demand for our drug candidates;
• injury to our reputation;
• withdrawal of clinical trial participants;
• costs of related litigation;
• substantial monetary awards to patients and others;
• loss of revenues; and
• the inability to commercialize drug candidates.
With respect to product liability claims, we could face additional liability beyond insurance limits if testing mistakes were to endanger any human subjects. In addition, if a claim is made against us in conjunction with these research testing activities, the market price of our ADSs may be negatively affected.
The outbreak of the novel strain of coronavirus, SARS-CoV-2, which causes COVID-19, could adversely impact our business, including our non-clinical studies and clinical trials.
Public health crises such as pandemics or similar outbreaks might adversely impact our business. In December 2019, a novel strain of coronavirus, SARS-CoV-2, which causes coronavirus disease 2019 (COVID-19), surfaced in Wuhan, China. Since then, COVID-19 has spread to most countries in the world.
As a result of the COVID-19 outbreak, or similar pandemics, we have and may in the future experience disruptions that could severely impact our business, preclinical studies and clinical trials, including:
• delays or difficulties in enrolling patients in our clinical trials;
• delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff;
• delays or disruptions in non-clinical experiments and investigational new drug application-enabling good laboratory practice standard toxicology studies due to unforeseen circumstances at contract research organizations and vendors along their supply chain;
• increased rates of patients withdrawing from our clinical trials following enrollment as a result of contracting COVID-19, being forced to quarantine, or not wanting to attend hospital visits;
18
• diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials;
• interruption of key clinical trial activities, such as clinical trial site data monitoring, due to limitations on travel imposed or recommended by national, state or local governments, employers and others or interruption of clinical trial subject visits and study procedures (particularly any procedures that may be deemed non-essential), which may impact the integrity of subject data and clinical study endpoints;
• interruption or delays in the operations of the U.S. Food and Drug Administration, the European Medicines Agency, the Australian Therapeutic Goods Administration or other foreign regulatory agencies, which may impact approval timelines;
• interruption of, or delays in receiving, supplies of our drug candidates from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in our supply chain or distribution vendors’ ability to ship drug candidates; and
• limitations on employee resources that would otherwise be focused on the conduct of our non-clinical studies and clinical trials, including because of sickness of employees or their families, the desire of employees to avoid contact with large groups of people, an increased reliance on working from home or mass transit disruptions.
Product shipment delays could have a material adverse effect on our business, results of operations and financial condition.
The shipment, import and export of our drug candidates and the API used to manufacture them will require import and export licenses. In the United States, the FDA, U.S. Customs and Border Protection, and the DEA; in Canada, the Canada Border Services Agency, Health Canada; in Europe, the EMA and the European Commission; in Australia and New Zealand, the Australian Customs and Board Protection Service, the TGA, the New Zealand Medicines and Medical Device Safety Authority and the New Zealand Customs Service; and in other countries, similar regulatory authorities, regulate the import and export of pharmaceutical products that contain controlled substances. Specifically, the import and export processes require the issuance of import and export licenses by the relevant controlled substance authority in both the importing and exporting country.
We may not be granted, or if granted, maintain, such licenses from the authorities in certain countries. Even if we obtain the relevant licenses, shipments of API and our drug candidates may be held up or lost in transit, which could cause significant delays and may lead to product batches being stored outside required temperature ranges. Inappropriate storage may damage the product shipment resulting in delays in clinical trials or, upon commercialization, a partial or total loss of revenue from one or more shipments of API or our drug candidates. A delay in a clinical trial or, upon commercialization, a partial or total loss of revenue from one or more shipments of API or our drug candidates could have a material adverse effect on our business, results of operations and financial condition.
Our drug candidates will be subject to controlled substance laws and regulations. Failure to receive necessary approvals may delay the launch of our drug candidates and failure to comply with these laws and regulations may adversely affect the results of our business operations.
Our drug candidates contain controlled substances as defined in the Controlled Substance Act (“CSA”). Controlled substances that are pharmaceutical products are subject to a high degree of regulation under the CSA, which establishes, among other things, certain registration, manufacturing quotas, security, recordkeeping, reporting, import, export and other requirements administered by the DEA. The DEA classifies controlled substances into five schedules: Schedule I, II, III, IV or V substances. Schedule I substances by definition have a high potential for abuse, have not currently “accepted medical use” in the United States, lack accepted safety for use under medical supervision, and may not be prescribed, marketed or sold in the United States. Pharmaceutical products approved for use in the United States may be listed as Schedule II, III, IV or V, with Schedule II substances considered to present the highest potential for abuse or dependence and Schedule V substances the lowest relative risk of abuse among such substances. Schedule I and II drugs are subject to the strictest controls under the CSA, including manufacturing and procurement quotas, security requirements and criteria for importation. In addition, dispensing of Schedule II drugs is further restricted. For example, they may not be refilled without a new prescription.
19
As a synthetic cannabinoids pharmaceutical product with psychedelic agents, our drug candidates are likely to be scheduled as Schedule II or III controlled substance. We will need to identify wholesale distributors with the appropriate DEA registrations and authority to distribute the products to pharmacies and other healthcare providers, and these distributors would need to obtain Schedule II or III distribution registrations. The failure to obtain, or delay in obtaining, or the loss of any of those registrations could result in increased costs to us. If any of our drug candidates is a Schedule II drug, pharmacies would have to maintain enhanced security with alarms and monitoring systems, and they must adhere to additional recordkeeping and inventory requirements. This may discourage some pharmacies from carrying the product. Furthermore, state and federal enforcement actions, regulatory requirements, and legislation intended to reduce prescription drug abuse, such as the requirement that physicians consult a state prescription drug monitoring program, may make physicians less willing to prescribe, and pharmacies to dispense, Schedule II products.
We intend to manufacture the commercial supply of our drug candidates outside of the United States. If any of our products are approved by the FDA and classified as a Schedule II or III substance, an importer can import for commercial purposes if it obtains from the DEA an importer registration and files an application with the DEA for an import permit for each import. The failure to identify an importer or obtain the necessary import authority could affect the availability of our drug candidates and have a material adverse effect on our business, results of operations and financial condition. In addition, an application for a Schedule II importer registration must be published in the Federal Register, and there is a waiting period for third party comments to be submitted. The failure to maintain the necessary registrations or comply with applicable laws could delay the commercialization of our drug candidates and could delay the completion of the clinical studies. Furthermore, failure to maintain compliance with the CSA, particularly non-compliance resulting in loss or diversion, could result in regulatory action that could have a material adverse effect on our business, financial condition and results of operations. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to restrict, suspend or revoke those registrations. In certain circumstances, violations could lead to criminal proceedings. In addition, if the FDA, DEA, or any foreign regulatory authority determines that our drug candidates may have potential for abuse, it may require us to generate more clinical or other data than we currently anticipate to establish whether or to what extent the substance has an abuse potential, which could increase the cost and/or delay the launch of which could increase the cost and/or delay the launch of our drug candidates.
Individual states have also established controlled substance laws and regulations. Though state-controlled substance laws often mirror federal law, because the states are separate jurisdictions, they may separately schedule our drug candidates as well. While some states automatically schedule a drug based on federal action, other states schedule drugs through rulemaking or a legislative action. State scheduling may delay commercial sale of any product for which we obtain federal regulatory approval and adverse scheduling could have a material adverse effect on the commercial attractiveness of such product. We or our partners must also obtain separate state registrations, permits or licenses in order to be able to obtain, handle, and distribute controlled substances for clinical trials or commercial sale, and failure to meet applicable regulatory requirements could lead to enforcement and sanctions by the states in addition to those from the DEA or otherwise arising under federal law.
We intend to contract manufacturers in Australia to produce the drug product for our clinical trials and the API for our drug candidates. In addition, we may decide to develop, manufacture or commercialize our drug candidates in additional countries. As a result, we will also be subject to controlled substance laws and regulations from the TGA in Australia and from other regulatory agencies in other countries where we develop, manufacture or commercialize our drug candidates in the future. We plan to submit NDAs for our drug candidates to the FDA upon completion of all requisite clinical trials and may require additional DEA scheduling decisions at such time as well.
Changes in U.S. healthcare law and implementing regulations, as well as changes in healthcare policy, may impact our business in ways that we cannot currently predict and may harm our business and results of operations.
There have been, and likely will continue to be, several executive, legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of drug candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any drug candidates for which we obtain marketing approval. Among policy makers and payors in the United States there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access and the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
20
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, collectively referred to as the Affordable Care Act, substantially changed the way healthcare is financed by both the government and private insurers, and significantly impacts the U.S. pharmaceutical industry. The Affordable Care Act, among other things: (1) introduced a new average manufacturer price definition for drugs and biologics that are inhaled, infused, instilled, implanted or injected and not generally dispensed through retail community pharmacies; (2) increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and expanded rebate liability from fee-for-service Medicaid utilization to include the utilization of Medicaid managed care organizations as well; (3) established a branded prescription drug fee that pharmaceutical manufacturers of branded prescription drugs must pay to the federal government; (4) expanded the list of covered entities eligible to participate in the 340B drug pricing program by adding new entities eligible for the program; (5) established a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% (increased to 70% pursuant to the Bipartisan Budget Act of 2018, or BBA, effective as of January 2019) point-of-sale discounts off negotiated prices of applicable branded drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; (6) extended manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; (7) expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals, including individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; (8) created a licensure framework for follow-on biologic products; and (9) established a Center for Medicare and Medicaid Innovation at the CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending.
There remain judicial and Congressional challenges to certain aspects of the Affordable Care Act, as well as efforts by the Trump administration to repeal or replace certain aspects of the Affordable Care Act. For example, the Tax Cuts and Jobs Act of 2017, or the TCJA, was enacted, which included a provision that repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the Affordable Care Act on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas ruled that the individual mandate is a critical and inseverable feature of the Affordable Care Act, and therefore, because it was repealed as part of the TCJA, the remaining provisions of the Affordable Care Act are invalid as well. On December 18, 2019, the U.S. Court of Appeals for the 5th Circuit ruled that the individual mandate was unconstitutional but remanded the case back to the District Court to determine whether the remaining provisions of the Affordable Care Act are invalid as well. On March 2, 2020, the U.S. Supreme Court granted the petitions for writs of certiorari to review the case, although it is unclear when a decision will be made or how the Supreme Court will rule. In addition, there may be other efforts to challenge, repeal or replace the Affordable Care Act. We are continuing to monitor any changes to the Affordable Care Act that, in turn, may potentially impact our business in the future.
Other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes include aggregate reductions to Medicare payments to providers of 2% per fiscal year pursuant to the Budget Control Act of 2011 and subsequent laws, which began in 2013 and will remain in effect through 2030, unless additional Congressional action is taken. These Medicare sequester reductions will be suspended from May 1, 2020 through December 31, 2021 due to the COVID-19 pandemic. In January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. New laws may result in additional reductions in Medicare and other healthcare funding, which may materially negatively affect customer demand and affordability for our drug candidates and, accordingly, the results of our financial operations. Also, there has been heightened governmental scrutiny recently over the manner in which pharmaceutical companies set prices for their marketed products, which have resulted in several Congressional inquiries and proposed federal legislation, as well as state efforts, designed to, among other things, bring more transparency to product pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. At the state level, individual states in the United States are increasingly active in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or
21
patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions on coverage or access could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our drug candidates that we successfully commercialize or put pressure on our product pricing.
In addition, proposed federal and state legislation may increase competition as it relates to cannabis derived products. Under the Cannabis Administration and Opportunity Act, the U.S. Senate proposed legalizing the use of hemp-derived CBD in dietary supplements by amending the FDCA. The Hemp Access and Consumer Safety Act of 2021 (SB 1698) also permits hemp-derived CBD to be used in dietary supplements. States are considering the reimbursement of medical marijuana. For example, New Jersey lawmakers introduced legislation, which is still pending, that requires reimbursement for medical marijuana under certain circumstances, while New York lawmakers introduced pending legislation that classifies medical marijuana as a prescription drug that may be covered for workers’ compensation purposes. As the availability and reimbursement of cannabis-derived products potentially expand, the pharmaceutical industry may directly compete with state-regulated cannabis businesses for market share.
We expect that these and other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and lower reimbursement, and put additional downward pressure on the price that we receive for any approved product. It is possible that additional governmental action is taken in response to the COVID-19 pandemic. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action in the United States. If we or any third parties we may engage are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our drug candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability
Risks Related to Intellectual Property
Our success depends on our ability to protect our intellectual property and our proprietary technology.
Our success is to a certain degree also dependent on our ability to obtain and maintain patent protection or where applicable, to receive/maintain orphan drug designation/status and resulting marketing exclusivity for our drug candidates.
We may be materially adversely affected by our failure or inability to protect our intellectual property rights. Without the granting of these rights, the ability to pursue damages for infringement would be limited. Similarly, any know-how that is proprietary or particular to our technologies may be subject to risk of disclosure by employees or consultants despite having confidentiality agreements in place.
Any future success will depend in part on whether we can obtain and maintain patents to protect our own products and technologies; obtain licenses to the patented technologies of third parties; and operate without infringing on the proprietary rights of third parties. Biotechnology patent matters can involve complex legal and scientific questions, and it is impossible to predict the outcome of biotechnology and pharmaceutical patent claims. Any of our future patent applications may not be approved, or we may not develop additional products or processes that are patentable. Some countries in which we may sell our drug candidate or license our intellectual property may fail to protect our intellectual property rights to the same extent as the protection that may be afforded in the United States or Australia. Some legal principles remain unresolved and there has not been a consistent policy regarding the breadth or interpretation of claims allowed in patents in the United States, the United Kingdom, the European Union, Australia or elsewhere. In addition, the specific content of patents and patent applications that are necessary to support and interpret patent claims is highly uncertain due to the complex nature of the relevant legal, scientific and factual issues. Changes in either patent laws or in interpretations of patent laws in the United States, the United Kingdom, the European Union or elsewhere may diminish the value of our intellectual property or narrow the scope of our patent protection. Even if we are able to obtain patents, they may not issue in a form that
22
will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner. We may also fail to take the required actions or pay the necessary fees to maintain our patents.
Moreover, any of our pending applications may be subject to a third party pre-issuance submission of prior art to the U.S. Patent and Trademark Office, or USPTO, the European Patent Office, or EPO, the Intellectual Property Office, or IPO, in the United Kingdom, the Australian Patent and Trademark Office and/or any patents issuing thereon may become involved in opposition, derivation, reexamination, inter partes review, post grant review, interference proceedings or other patent office proceedings or litigation, in the United States or elsewhere, challenging our patent rights. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, and allow third parties to commercialize our technology or products and compete directly with us, without payment to us. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to exploit our intellectual property or develop or commercialize current or future drug candidates.
The issuance of a patent is not conclusive as to the inventorship, scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the United States, the European Union, Australia and elsewhere. Such challenges may result in loss of ownership or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit the duration of the patent protection of our technology and products. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
In addition, other companies may attempt to circumvent any regulatory data protection or market exclusivity that we obtain under applicable legislation, which may require us to allocate significant resources to preventing such circumvention. Such developments could enable other companies to circumvent our intellectual property rights and use our clinical trial data to obtain marketing authorizations in the European Union, Australia and in other jurisdictions. Such developments may also require us to allocate significant resources to prevent other companies from circumventing or violating our intellectual property rights.
Intellectual property rights of third parties could adversely affect our ability to commercialize our drug candidates, such that we could be required to litigate with or obtain licenses from third parties in order to develop or market our drug candidates.
Our commercial success may depend upon our future ability and the ability of our potential collaborators to develop, manufacture, market and sell our drug candidates without infringing valid intellectual property rights of third parties.
If a third-party intellectual property right exists it may require the pursuit of litigation or administrative proceedings to nullify or invalidate the third party intellectual property right concerned, or entry into a license agreement with the intellectual property right holder, which may not be available on commercially reasonable terms, if at all.
Third-party intellectual property right holders, including our competitors, may bring infringement claims against us. We may not be able to successfully settle or otherwise resolve such infringement claims. If we are unable to successfully settle future claims or otherwise resolve such claims on terms acceptable to us, we may be required to engage in or continue costly, unpredictable and time-consuming litigation and may be prevented from, or experience substantial delays in, marketing our drug candidate.
If we fail to settle or otherwise resolve any such dispute, in addition to being forced to pay damages, we or our potential collaborators may be prohibited from commercializing any drug candidates we may develop that are held to be infringing, for the duration of the patent term. We might, if possible, also be forced to redesign our formulations so that we no longer infringe such third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
23
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Because we collaborate with various organizations and academic institutions on the advancement of our technology and drug candidates, we may, at times, share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, such as trade secrets. Despite these contractual provisions, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by potential competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, discovery by a third party of our trade secrets or other unauthorized use or disclosure would impair our intellectual property rights and protections in our drug candidates.
In addition, these agreements typically restrict the ability of our collaborators, advisors, employees and consultants to publish data potentially relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. In other cases, publication rights are controlled exclusively by us. In other cases, we may share these rights with other parties. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications are required to be paid to the United State Patent and Trademark Office and other governmental patent agencies outside of the United States in several stages over the lifetime of the patents and applications. The USPTO and various corresponding governmental patent agencies outside of the United States require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process and after a patent has issued. There are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction.
We may become involved in lawsuits to protect and defend our patents or other intellectual property, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or other intellectual property and we may inadvertently infringe the patent or intellectual property of others. To counter infringement or unauthorized use, we may be required to file claims, and any related litigation and/or prosecution of such claims can be expensive and time consuming. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their intellectual property. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid in whole or in part, unenforceable, or construe the patent’s claims narrowly allowing the other party to commercialize competing products on the grounds that our patents do not cover such products.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our technical and management personnel from their normal responsibilities. Such litigation or proceedings could substantially increase our operating losses and reduce our resources available for development activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. The effects of patent litigation or other proceedings could therefore have a material adverse effect on our ability to compete in the marketplace.
24
Confidentiality and invention assignment agreements with our employees, advisors and consultants may not adequately prevent disclosure of trade secrets and protect other proprietary information.
We consider proprietary trade secrets and/or confidential know-how and unpatented know-how to be important to our business. We may rely on trade secrets and/or confidential know-how to protect our technology, especially where patent protection is believed by us to be of limited value. However, trade secrets and/or confidential know-how can be difficult to maintain as confidential.
To protect this type of information against disclosure or appropriation by competitors, our policy is to require our employees, advisors and consultants to enter into confidentiality and invention assignment agreements with us. However, current or former employees, advisors and consultants may unintentionally or willfully disclose our confidential information to competitors, and confidentiality and invention assignment agreements may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Enforcing a claim that a third party obtained illegally and is using trade secrets and/or confidential know-how is expensive, time consuming and unpredictable. The enforceability of confidentiality and invention assignment agreements may vary from jurisdiction to jurisdiction.
Failure to obtain or maintain trade secrets and/or confidential know-how trade protection could adversely affect our competitive position. Moreover, our competitors may independently develop substantially equivalent proprietary information and may even apply for patent protection in respect of the same. If successful in obtaining such patent protection, our competitors could limit our use of our trade secrets and/or confidential know-how.
Intellectual property rights do not address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
• Others may be able to make products that are similar to ours but that are not covered by our intellectual property rights.
• Others may independently develop similar or alternative technologies or otherwise circumvent any of our technologies without infringing our intellectual property rights.
• We or any of our collaboration partners might not have been the first to conceive and reduce to practice the inventions covered by the patents or patent applications that we own, license or will own or license.
• We or any of our collaboration partners might not have been the first to file patent applications covering certain of the patents or patent applications that we or they own or have obtained a license, or will own or will have obtained a license.
• It is possible that any pending patent applications that we have filed, or will file, will not lead to issued patents.
• Issued patents that we own may not provide us with any competitive advantage, or may be held invalid or unenforceable, as a result of legal challenges by our competitors.
• Our competitors might conduct research and development activities in countries where we do not have patent rights, or in countries where research and development safe harbor laws exist, and then use the information learned from such activities to develop competitive products for sale in our major commercial markets.
• Ownership of our patents or patent applications may be challenged by third parties.
• The patents of third parties or pending or future applications of third parties, if issued, may have an adverse effect on our business.
25
We may face difficulties with protecting our intellectual property in certain jurisdictions, which may diminish the value of our intellectual property rights in those jurisdictions.
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws in the United States and the European Union, and many companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. If we or our collaboration partners encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished and we may face additional competition from others in those jurisdictions.
Some countries in Europe and China have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we are, or any of our licensors is, forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position or commercial advantage may be impaired and our business and results of operations may be adversely affected.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our drug candidates and any future drug candidates.
As is the case with other biotechnology and pharmaceutical companies, our success is heavily dependent on intellectual property rights, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves technological and legal complexity, and obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. The U.S. Supreme Court in recent years has issued rulings either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations or ruling that certain subject matter is not eligible for patent protection. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by Congress, the federal courts, the USPTO and equivalent bodies in non-U.S. jurisdictions, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce existing patents and patents we may obtain in the future.
Patent reform laws, such as the Leahy-Smith America Invents Act, or the Leahy-Smith Act, as well as changes in how patent laws are interpreted, could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. The Leahy-Smith Act made a number of significant changes to U.S. patent law. These include provisions that affect the filing and prosecution strategies associated with patent applications, including a change from a “first-to-invent” to a “first-inventor-to-file” patent system, and a change allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO-administered post-grant proceedings, including post-grant review, inter partes review and derivation proceedings. The USPTO has developed regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act and, in particular, the “first-inventor-to-file” provisions, became effective in 2013. The Leahy-Smith Act and its implementation may increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have harm our business, financial condition and results of operations.
Price controls may be imposed in non-U.S. markets, which may negatively affect our future profitability.
In some countries, particularly EU member states, Japan, Australia and Canada, the pricing of prescription drugs is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of marketing approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various EU member states and parallel distribution, or arbitrage between low-priced and
26
high-priced member states, can further reduce prices. In some countries, we or our collaborators may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our drug candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of our drug candidates is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business, revenues or profitability could be harmed.
Risks Relating to Ownership of the ADSs and this Offering
The trading price of the ADSs may be volatile, and purchasers of the ADSs could incur substantial losses.
The market price of our ordinary shares historically has been, and we expect our ordinary shares and ADSs will continue to be, subject to significant fluctuations over short periods of time. These fluctuations may be due to factors specific to us, to changes in analysts’ recommendations and earnings estimates, to arbitrage between our Australian-listed ordinary shares and our Nasdaq-listed ADSs, to changes in exchange rates, or to factors affecting the biopharmaceutical industry or the securities markets in general. Market fluctuations, as well as general political and economic conditions, such as a recession, interest rate or currency fluctuations, could adversely affect the market price of our securities.
We may experience a material decline in the market price of our shares, regardless of our operating performance. Therefore, a holder of our ADSs may not be able to sell those ADSs at or above the price paid by such holder for such ADSs. Price declines in our ADSs could result from a variety of factors, including many outside our control. These factors include:
• the results of pre-clinical testing and clinical trials by us and our competitors;
• unforeseen safety issues or adverse side effects resulting from the clinical trials or the commercial use of our drug candidate;
• regulatory actions in respect of any of our drug candidates or the products of any of our competitors;
• announcements of the introduction of new products by us or our competitors;
• market conditions, including market conditions in the pharmaceutical and biotechnology sectors;
• increases in our costs or decreases in our revenues due to unfavorable movements in foreign currency exchange rates;
• developments or litigation concerning patents, licenses and other intellectual property rights;
• litigation or public concern about the safety of our drug candidates;
• changes in recommendations or earnings estimates by securities analysts;
• actual and anticipated fluctuations in our quarterly operating results;
• deviations in our operating results from the estimates of securities analysts;
• rumors relating to us or our competitors;
• additions or departures of key personnel;
• changes in third party reimbursement policies; and
• developments concerning current or future strategic alliances or acquisitions.
In addition, volatility and low market price of our ADSs may adversely impact investors’ interest in our securities. A decline in investors’ interest may prompt further volatility and decrease in market price.
27
There has been no prior market for the ADSs and an active and liquid market for our securities may fail to develop, which could harm the market price of the ADSs.
While our ordinary shares have been listed on the Australian Securities Exchange, or ASX, prior to this offering, there has been no public market on a U.S. national securities exchange for our ordinary shares or ADSs. Although we have applied for the listing of the ADSs on Nasdaq, an active trading market for the ADSs may never develop or be sustained following this offering. The initial offering price of the ADSs will be determined through negotiations between us and the underwriter and will be based, in part, on prevailing market prices of our ordinary shares on the ASX, after taking into account market conditions and other factors. This offering price may not be indicative of the market price of the ADSs after this offering. In the absence of an active trading market for the ADSs, investors may not be able to sell their ADSs at or above the offering price or at the time that they would like to sell.
If we are or become a passive foreign investment company (“PFIC” ), then that would subject our U.S. investors to adverse tax rules.
Holders of our ADSs who are U.S. taxpayers will be subject to particular income tax rules if we are a passive foreign investment company, or PFIC. These rules could result in a reduction in the after-tax return to a “U.S. Holder” of our ADSs and reduce the value of our ADSs. For U.S. federal income tax purposes, we will be classified as a PFIC for any taxable year in which (i) 75% or more of our gross income is passive income, or (ii) at least 50% of the average value of all of our assets for the taxable year produce or are held for the production of passive income. For this purpose, cash is considered to be an asset that produces passive income.
If we are classified as a PFIC in any year that a U.S. Holder owns ADSs, the U.S. Holder will generally continue to be treated as holding ADSs of a PFIC in all subsequent years, notwithstanding that we are not classified as a PFIC in a subsequent year. Dividends received by the U.S. Holder and gains realized from the sale of our ADSs would be taxed as ordinary income and subject to an interest charge. We urge U.S. investors to consult their own tax advisors about the application of the PFIC rules and certain elections that may help to minimize adverse U.S. federal income tax consequences in their particular circumstances.
The requirements of being a public company may strain our resources and divert management’s attention and if we are unable to maintain effective internal control over financial reporting in the future, the accuracy and timeliness of our financial reporting may be adversely affected.
As a U.S. publicly-traded company, we will be subject to the reporting requirements of the U.S. Securities Exchange Act of 1934, as amended (the Exchange Act), the Sarbanes-Oxley Act, the Dodd-Frank Act, the listing requirements of the Nasdaq Capital Market and other applicable securities rules and regulations. Compliance with these rules and regulations will increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and increase demand on our systems and resources. The Exchange Act requires, among other things, that we file certain reports with respect to our business and results of operations. The Sarbanes-Oxley Act requires that we maintain effective disclosure controls and procedures and internal control over financial reporting. In order to maintain and, if required, improve our disclosure controls and procedures and internal control over financial reporting to meet this standard, significant resources and management oversight are required. As a result, management’s attention may be diverted from other business concerns, which could adversely affect our business and results of operations.
The Sarbanes-Oxley Act requires that we assess the effectiveness of our internal control over financial reporting annually and disclosure controls and procedures quarterly. If we identify material weaknesses in future periods or we are not able to comply with the requirements of Section 404 in a timely manner, our reported financial results could be restated, we could be subject to investigations or sanctions by regulatory authorities, which would require additional financial and management resources, and the market price of our ordinary shares could decline.
You will experience immediate and substantial dilution in the net tangible book value of the ADSs you purchase in this offering.
The initial public offering price of the ADSs is substantially higher than the net tangible book value per ADS or per ordinary share immediately after this offering. If you purchase ADSs in this offering, you will suffer immediate dilution of US$ per ADS (or US$ per ordinary share), or US$ per ADS
28
(or US$ per ordinary share) if the underwriter exercises its option to purchase additional shares in full, representing the difference between our as adjusted net tangible book value per ADS or per ordinary share after giving effect to the sale of ADSs in this offering and the initial public offering price of US$ per ADS. See “Dilution.”
Our issuance of additional ordinary shares in connection with financings, acquisitions, investments, or otherwise will dilute all other ADS holders.
We expect to issue additional ordinary shares in the future that will result in dilution to all other ADS holders. We may also raise capital through equity financings in the future. As part of our business strategy, we may acquire or make investments in complementary companies, products, or technologies and issue equity securities to pay for any such acquisition or investment. While we will be subject to the constraints of the ASX Listing Rules regarding the percentage of our capital that we are able to issue within a 12-month period (subject to applicable exceptions), any such issuances of additional ordinary shares may cause ADS holders to experience significant dilution of their ownership interests and the per ADS value of our ADSs to decline.
As long as we remain subject to the rules of the ASX and Nasdaq, we will be unable to access equity capital without shareholder approval if such equity capital sales would result in an equity issuance above regulatory thresholds and, consequently, we could be unable to obtain financing sufficient to sustain our business if we are unsuccessful in soliciting requisite shareholder approvals.
Our ability to access equity capital is currently limited by ASX Listing Rule 7.1, which provides that a company must not, subject to specified exceptions (including approval by shareholders), issue or agree to issue during any consecutive 12-month period any equity securities, or other securities with rights to conversion to equity, if the number of those securities in aggregate would exceed 15% of the number of ordinary securities on issue at the commencement of that 12-month period.
Our equity issuances will be limited by ASX Listing Rule 7.1 as long as we continue to be listed on the ASX and this constraint may prevent us from raising the full amount of equity capital needed for operations without prior shareholder approval.
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
We will have broad discretion in the application of the net proceeds that we receive from this offering as well as of our existing cash, and we may spend or invest these funds in a way with which our shareholders or holders of the ADSs disagree. Our failure to apply these funds effectively could harm our business and financial condition. Pending their use, we may invest the net proceeds from the offering in a manner that does not produce income or that loses value. These investments may not yield a favorable return to our investors.
Future sales of ordinary shares or ADSs by existing holders could depress the market price of the ordinary shares or ADSs.
Based on 1,068,411,224 ordinary shares outstanding as of June 30, 2021, upon the closing of this offering, we will have outstanding a total of ordinary shares (including ordinary shares represented by ADSs), assuming no exercise of the underwriters’ option to purchase additional ADSs and no exercise of outstanding options warrants offered and sold in this offering. Each member of our senior management and board of directors and their affiliates are subject to lock-up agreements with the underwriters that restrict their ability to transfer ordinary shares, options and other securities convertible into, exchangeable for, or exercisable for ordinary shares during the period ending on, and including, the 180th day after the date of this prospectus, subject to specified exceptions. Roth Capital Partners, LLC may, in its sole discretion, permit our shareholders who are subject to these lock-up agreements to sell securities prior to the expiration of the lock-up agreements. As of the date of this prospectus, the exercise of all outstanding options exercisable for ordinary shares would enable the subscription of new ordinary shares representing approximately of the diluted share capital.
After the lock-up agreements pertaining to this offering expire, 94,302,045 additional ordinary shares will be eligible for sale in the public market, all of which ordinary shares are held by members of our senior management and board of directors and will be subject to volume limitations under Rule 144 under the Securities Act of 1933,
29
as amended, or the Securities Act. In addition, the ordinary shares subject to subscription under outstanding options exercisable for ordinary shares will become eligible for sale in the public market in the future, subject to certain legal and contractual limitations. Sales of a large number of the ordinary shares in the public market could depress the market price of the ADSs. See “Shares and American Depository Shares Eligible for Future Sale” for a more detailed description of sales that may occur in the future. If these additional ordinary shares are sold, or if it is perceived that they will be sold, in the public market, the trading price of the ordinary shares and ADSs could decline substantially, which could impair our ability to raise additional capital through the issuance of ordinary shares, ADSs or other securities in the future.
The dual listing of our ordinary shares and the ADSs following this offering may negatively impact the liquidity and value of the ADSs.
Following this offering and after the ADSs are listed on Nasdaq, our ordinary shares will continue to be listed on the ASX. We cannot predict the effect of this dual listing on the value of our ordinary shares and ADSs. However, the dual listing of our ordinary shares and ADSs may dilute the liquidity of these securities in one or both markets and may negatively impact the development of an active trading market for the ADSs in the United States. The price of the ADSs could also be negatively impacted by trading in our ordinary shares on the ASX.
We are subject to risks associated with currency fluctuations, and changes in foreign currency exchange rates could impact our results of operations.
Our ordinary shares are quoted in Australian dollars on the ASX and the ADSs will be quoted in U.S. dollars. In the past year, the Australian dollar has generally weakened against the U.S. dollar; however, this trend may not continue and may be reversed. As such, any significant change in the value of the Australian dollar may have a negative effect on the value of the ADSs in U.S. dollars. In addition, if the Australian dollar weakens against the U.S. dollar, then, if we decide to convert our Australian dollars into U.S. dollars for any business purpose, appreciation of the U.S. dollar against the Australian dollar would have a negative effect on the U.S. dollar amount available to us. To the extent that we need to convert U.S. dollars we receive from this offering into Australian dollars for our operations, appreciation of the Australian dollar against the U.S. dollar would have a negative effect on the Australian dollar amount we would receive from the conversion. Consequently, appreciation or depreciation in the value of the Australian dollar relative to the U.S. dollar would affect our financial results reported in U.S. dollar terms without giving effect to any underlying change in our business or results of operations. As a result of such foreign currency fluctuations, it could be more difficult to detect underlying trends in our business and results of operations.
Our ADS holders are not shareholders and do not have shareholder rights.
Deutsche Bank, as depositary, registers and delivers our American Depositary Shares, or ADSs. Our ADS holders will not be treated as shareholders and do not have the rights of shareholders. The depositary will be the holder of the shares underlying our ADSs. Holders of our ADSs will have ADS holder rights. A deposit agreement among us, the depositary and our ADS holders, and the beneficial owners of ADSs, sets out ADS holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and the ADSs. For a description of ADS holder rights, see “Description of American Depositary Shares” in this Registration Statement.
Our shareholders have shareholder rights. Australian law and our constitution govern shareholder rights. For a description of our shareholders’ rights, see “Memorandum and Articles of Association” in this Registration Statement. Shareholders are entitled to our notices of general meetings and to attend and vote at our general meetings of shareholders. At a general meeting, every shareholder present (in person or by proxy, attorney or representative) and entitled to vote has one vote on a show of hands. Every shareholder present (in person or by proxy, attorney or representative) and entitled to vote has one vote per fully paid ordinary share on a poll. This is subject to any other rights or restrictions which may be attached to any shares.
Our ADS holders do not have the same voting rights as our shareholders. ADS holders may exercise voting rights with respect to the underlying ordinary shares only in accordance with the provisions of the deposit agreement. Under the deposit agreement, ADS holders vote by giving voting instructions to the depositary. Upon receipt of instructions, the depositary will try to vote in accordance with those instructions. Otherwise, ADS holders will not be able to vote unless they withdraw the ordinary shares underlying their ADSs.
30
If we ask for our ADS holders’ instructions, the depositary will notify our ADS holders of the upcoming vote and arrange to deliver our voting materials and form of notice to them. The depositary will try, as far as practical, subject to Australian law and the provisions of the depositary agreement, to vote the shares as our ADS holders instruct. The depositary will not vote or attempt to exercise the right to vote other than in accordance with the instructions of the ADS holders. We cannot assure our ADS holders that they will learn of ordinary shareholders’ meetings and receive the voting materials in time to instruct the depositary or withdraw the underlying ordinary shares. This means that there is a risk that our ADS holders may not be able to exercise voting rights and there may be nothing they can do if their shares are not voted as they requested.
Our ADS holders do not have the same rights to receive dividends or other distributions as our shareholders.
Subject to any special rights or restrictions attached to a share, the directors may determine that a dividend will be payable on a share and fix the amount, the time for payment and the method for payment (although we have never declared or paid any cash dividends on our ordinary shares and we do not anticipate paying any cash dividends in the foreseeable future). Dividends may be paid on shares of one class but not another and at different rates for different classes. Dividends and other distributions payable to our shareholders with respect to our ordinary shares generally will be payable directly to them. Any dividends or distributions payable with respect to ordinary shares will be paid to the depositary, which has agreed to pay to our ADS holders the cash dividends or other distributions it or the custodian receives on shares or other deposited securities, after deducting its fees and expenses. Our ADS holders will receive these distributions in proportion to the number of shares their ADSs represent. In addition, there may be certain circumstances in which the depositary may not pay to our ADS holders amounts distributed by us as a dividend or distribution.
There are circumstances where it may be unlawful or impractical to make distributions to the holders of our ADSs.
The deposit agreement with the depositary generally requires the depositary to convert foreign currency it receives in respect of deposited securities into U.S. dollars and distribute the U.S. dollars to ADS holders, provided the depositary can do so on a reasonable basis. If it does not convert foreign currency, the depositary may distribute the foreign currency only to those ADS holders to whom it is possible to do so. If a distribution is payable by us in Australian dollars, the depositary will hold the foreign currency it cannot convert for the account of the ADS holders who have not been paid. It will not invest the foreign currency and it will not be liable for any interest. If the exchange rates fluctuate during a time when the depositary cannot convert the foreign currency, our ADS holders may lose some of the value of the distribution. The depositary is not responsible if it decides that it is unlawful or impractical to make a distribution available to any ADS holders. This means that our ADS holders may not receive the distributions we make on our shares or any value for them if it is illegal or impractical for us to make them available.
ADSs holders may not be entitled to a jury trial with respect to claims arising under the deposit agreement, which could augur less favorable results to the plaintiff(s) in any such action.
The deposit agreement governing the ADSs representing our ordinary shares provides that holders and beneficial owners of ADSs irrevocably waive the right to a trial by jury in any legal proceeding arising out of or relating to the deposit agreement or the ADSs against us or the depositary to the fullest extent permitted by applicable law. If this jury trial waiver provision is prohibited by applicable law, an action could nevertheless proceed under the terms of the deposit agreement with a jury trial. To our knowledge, the enforceability of a jury trial waiver under the federal securities laws has not been finally adjudicated by a federal court. However, we believe that a jury trial waiver provision is generally enforceable under the laws of the State of New York, which govern the deposit agreement, by a court of the State of New York or a federal court, which have non-exclusive jurisdiction over matters arising under the deposit agreement, applying such law.
In determining whether to enforce a jury trial waiver provision, New York courts and federal courts will consider whether the visibility of the jury trial waiver provision within the agreement is sufficiently prominent such that a party has knowingly waived any right to trial by jury. We believe that this is the case with respect to the deposit agreement and the ADSs. In addition, New York courts will not enforce a jury trial waiver provision in order to bar a viable setoff or counterclaim sounding in fraud or one which is based upon a creditor’s negligence in failing to liquidate collateral upon a guarantor’s demand, or in the case of an intentional tort claim (as opposed to a contract dispute), none of which we believe are applicable in the case of the deposit agreement or the ADSs. No condition, stipulation or provision of the deposit agreement or ADSs serves as a waiver by any holder or beneficial owner of ADSs or by us or the Depositary of compliance with any provision of the federal securities laws. If you or any other
31
holder or beneficial owner of ADSs brings a claim against us or the Depositary in connection with matters arising under the deposit agreement or the ADSs, you or such other holder or beneficial owner may not be entitled to a jury trial with respect to such claims, which may have the effect of limiting and discouraging lawsuits against us and/or the Depositary.
If a lawsuit is brought against us and/or the Depositary under the deposit agreement, it may be heard only by a judge or justice of the applicable trial court, which would be conducted according to different civil procedures and may determine different results than a trial by jury would have had, including results that could be less favorable to the plaintiff(s) in any such action, depending on, among other things, the nature of the claims, the judge or justice hearing such claims, and the venue of the hearing.
As the jury trial waiver relates to claims arising out of or relating to the ADSs or the deposit agreement, we believe that the waiver would likely continue to apply to purchasers of ADSs in secondary transactions. In addition, we believe that the waiver would likely continue to apply to ADS holders or beneficial owners who withdraw the ordinary shares from the ADS facility with respect to claims arising before the cancellation of the ADSs and the withdrawal of the ordinary shares, and the waiver would likely not apply to ADS holders or beneficial owners who subsequently withdraw the ordinary shares represented by ADSs from the ADS facility with respect to claims arising after the withdrawal. However, to our knowledge, there has been no case law on the applicability of the jury trial waiver to ADS holders or beneficial owners who withdraw the ordinary shares represented by the ADSs from the ADS facility. Nevertheless, if this jury trial waiver is not permitted by applicable law, an action could proceed under the terms of the deposit agreement with a jury trial. No condition, stipulation or provision of the deposit agreement or the ADSs serves as a waiver by any owner or holder of ADSs or by us or the Depositary of compliance with any substantive provision of the U.S. federal securities laws and the rules and regulations promulgated thereunder.
Risks Relating to Our Location in Australia
Australian takeovers laws may discourage takeover offers being made for us or may discourage the acquisition of large numbers of our shares.
We are incorporated in Australia and are subject to the takeover laws of Australia. Amongst other things, we are subject to the Corporations Act 2001 (Commonwealth of Australia). Subject to a range of exceptions, the Corporations Act prohibits the acquisition of a direct or indirect interest in our issued voting shares (including through the acquisition of ADSs) if the acquisition of that interest will lead to a person’s or someone else’s voting power in us increasing from 20% or below to more than 20% or increasing from a starting point that is above 20% and below 90%. Exceptions to the general prohibition include circumstances where the person makes a formal takeover bid for us, if the person obtains shareholder approval for the acquisition or if the person acquires less than 3% of the voting power of us in any rolling six-month period. Australian takeovers laws may discourage takeover offers being made for us or may discourage the acquisition of large numbers of our shares.
Holders of our ordinary shares or ADSs may have difficulty in effecting service of process in the United States or enforcing judgments obtained in the United States.
Holders of our ordinary shares or ADSs may have difficulties enforcing, in actions brought in courts in jurisdictions located outside the U.S., liabilities under U.S. securities laws. In particular, if such a holder sought to bring proceedings in Australia based on U.S. securities laws, the Australian court might consider:
• that it did not have jurisdiction; and/or
• that it was not an appropriate forum for such proceedings; and/or
• that, applying Australian conflict of laws rule, U.S. law (including U.S. securities laws) did not apply to the relationship between holders of our ordinary shares or ADSs and us or our directors and officers; and/or
• that the U.S. securities laws were of a public or penal nature and should not be enforced by the Australian court.
32
Holders of our ordinary shares and ADSs may also have difficulties enforcing in courts outside the U.S. judgments obtained in the U.S. courts against any of our directors and executive officers or us, including actions under the civil liability provisions of the U.S. securities laws.
As a foreign private issuer whose shares are listed on the Nasdaq Capital Market, we may follow certain home country corporate governance practices instead of certain Nasdaq requirements.
As a foreign private issuer whose shares are listed on the Nasdaq Capital Market, we are permitted to follow certain home country corporate governance practices instead of certain requirements of The Nasdaq Marketplace Rules. As an Australian company listed on the Nasdaq Capital Market, we may follow home country practice with regard to, among other things, the composition of the board of directors, director nomination process, compensation of officers and quorum at shareholders’ meetings. In addition, we may follow Australian law instead of the Nasdaq Marketplace Rules that require that we obtain shareholder approval for certain dilutive events, such as for the establishment or amendment of certain equity based compensation plans, an issuance that will result in a change of control of the company, certain transactions other than a public offering involving issuances of a 20% or more interest in the company and certain acquisitions of the shares or assets of another company. As a foreign private issuer that has elected to follow a home country practice instead of Nasdaq requirements. Accordingly, our shareholders may not be afforded the same protection as provided under Nasdaq’s corporate governance rules that are applicable to U.S. companies.
As a foreign private issuer, we are exempt from a number of rules under the U.S. securities laws and are permitted to file less information with the SEC than a U.S. company.
We are a foreign private issuer (as defined in the SEC’s rules) and, consequently, we are not subject to all of the disclosure requirements applicable to public companies organized within the United States. For example, we are exempt from certain rules under the Exchange Act that regulate disclosure obligations and procedural requirements related to the solicitation of proxies under the Exchange Act. In addition, our senior management and directors are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, while we currently make annual and semi-annual filings with respect to our listing on the ASX and expect to file financial reports on an annual and semi-annual basis, we will not be required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. public companies and will not be required to file quarterly reports on Form 10-Q or current reports on Form 8-K under the Exchange Act. In addition, foreign private issuers are not required to file their annual report on Form 20-F until four months after the end of each fiscal year. Accordingly, there may be less publicly available information concerning our company than there would be if we were not a foreign private issuer.
Any loss of our foreign private issuer status in the future could result in significant additional cost.
While we currently qualify as a foreign private issuer, the determination of foreign private issuer status is made annually on the last business day of an issuer’s most recently completed second fiscal quarter. In the future, we would lose our foreign private issuer status if we to fail to meet the requirements necessary to maintain our foreign private issuer status as of the relevant determination date. For example, if 50% or more of our securities are held by U.S. residents and more than 50% of our senior management or directors are residents or citizens of the United States, we could lose our foreign private issuer status.
The regulatory and compliance costs to us under U.S. securities laws as a U.S. domestic issuer could be significantly more than costs we incur as a foreign private issuer. If we were to cease to be a foreign private issuer, then we would be required to file periodic reports and registration statements on U.S. domestic issuer forms with the SEC, which forms are more detailed and extensive in certain respects than the forms available to a foreign private issuer. We would be required to prepare our financial statements in accordance with U.S. GAAP rather than IFRS. Such conversion of our financial statements to U.S. GAAP would involve significant time and cost. In addition, we may lose our ability to rely upon exemptions from certain corporate governance requirements on U.S. stock exchanges that are available to foreign private issuers such as the ones described above and exemptions from procedural requirements related to the solicitation of proxies.
33
U.S. investors may have difficulty enforcing civil liabilities against our company, our directors or members of senior management and the experts named in this prospectus.
Certain members of our senior management and board of directors named in this prospectus are non-residents of the United States, and a substantial portion of the assets of such persons are located outside the United States. As a result, it may be impracticable to serve process on such persons in the United States or to enforce judgments obtained in U.S. courts against them based on civil liability provisions of the securities laws of the United States. Even if you are successful in bringing such an action, there is doubt as to whether Australian courts would enforce certain civil liabilities under U.S. securities laws in original actions or judgments of U.S. courts based upon these civil liability provisions. In addition, awards of punitive damages in actions brought in the United States or elsewhere may be unenforceable in Australia or elsewhere outside the United States. An award for monetary damages under U.S. securities laws would be considered punitive if it does not seek to compensate the claimant for loss or damage suffered and is intended to punish the defendant. The enforceability of any judgment in Australia will depend on the particular facts of the case as well as the laws and treaties in effect at the time. The United States and Australia do not currently have a treaty or statute providing for recognition and enforcement of the judgments of the other country (other than arbitration awards) in civil and commercial matters.
As a result, our public shareholders may have more difficulty in protecting their interests through actions against us, our management or our directors than would shareholders of a corporation incorporated in a jurisdiction in the United States. In addition, as a company incorporated in Australia, the provisions of the Australian Corporations Act 2001 regulate the circumstances in which shareholder derivative actions may be commenced which may be different, and in many ways less permissive, than for companies incorporated in the United States.
34
This prospectus contains estimates and information concerning our industry and our business, including estimated market size and projected growth rates of the markets for our drug candidates. Unless otherwise expressly stated, we obtained this industry, business, market, medical and other information from reports, research surveys, studies and similar data prepared by third parties, industry, medical and general publications, government data and similar sources.
This information involves a number of assumptions and limitations. Although we are responsible for all of the disclosure contained in this prospectus and we believe the third-party market position, market opportunity and market size data included in this prospectus are reliable, we have not independently verified the accuracy or completeness of this third-party data. In addition, projections, assumptions and estimates of our future performance and the future performance of the industry in which we operate are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in “Risk Factors.” These and other factors could cause results to differ materially from those expressed in these publications and reports.
35
We estimate that the net proceeds to us from the sale of ADSs in this offering will be approximately US$ million (or approximately US$ million if the underwriter exercises its option to purchase additional ADSs in full), based on the assumed initial public offering price of US$ per ADS, after giving effect to, the ADS-to-ordinary share ratio of 1-to-50, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
Each US$1.00 increase (decrease) in the assumed initial offering price of US$ per ADS, after giving effect to the ADS-to-ordinary share ratio of 1-to-50, would increase (decrease) the net proceeds to us from this offering by approximately US$ million, assuming the number of ADSs offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, an increase (decrease) of 1,000,000 ADSs offered by us would increase (decrease) the net proceeds to us by US$ million, assuming the assumed initial public offering price of US$ per ADS, after giving effect to the ADS-to-ordinary share ratio of 1-to-50, remains the same and after deducting underwriting discounts and commissions.
We expect to use the net proceeds from this offering, together with our existing cash, to further our clinical trials, for working capital and other general corporate purposes. However, due to the uncertainties inherent in the product development process, it is difficult to estimate with certainty the exact amounts of the net proceeds from this offering that may be used for the above purposes. Our management will have broad discretion over the use of the net proceeds from this offering. The amounts and timing of our expenditures will depend upon numerous factors including the results of our research and development efforts, the timing and success of preclinical studies and any ongoing clinical trials or clinical trials we may commence in the future, the timing of regulatory submissions and the amount of cash obtained through future collaborations, if any.
We believe opportunities may exist from time to time to expand our current business through acquisitions or in-licensing of complementary companies, medicines or technologies. While we have no current agreements, commitments or understandings for any specific acquisitions or in-licensing at this time, we may use a portion of the net proceeds for these purposes.
As of December 31, 2020, we had cash of A$11,840,308 (or US$9,119,405). We believe our cash, together with the net proceeds of this offering, will be sufficient to fund our operations until . In particular, we estimate that such funds, together with such existing cash, will be sufficient to enable us to advance our clinical trials to the point where all programs have completed pivotal Phase 2 trials or two programs have completed pivotal Phase 2 trials and one program completed a Phase 3 clinical trial. The specifics of which trials are progressed first will depend on clinical success. At this stage we anticipate that the OSA, RA and IBD will advance the quickest. Funds for additional Phase 3 trials will be raised through future capital raises. It is estimated that completion of all six clinical development programs will cost approximately A$200 million.
Pending our use of the net proceeds from this offering, we intend to invest the net proceeds in a variety of capital preservation investments, including short-term, investment-grade, interest-bearing instruments.
36
We have never declared or paid any dividends on our ordinary shares. We intend to retain any earnings for use in our business and do not currently intend to pay cash dividends on our ordinary shares. Dividends, if any, on our outstanding ordinary shares will be declared by and subject to the discretion of our board of directors, and subject to Australian law.
Any dividend we declare will be paid to the holders of ADSs, subject to the terms of the deposit agreement, to the same extent as holders of our ordinary shares, to the extent permitted by applicable law and regulations, less the fees and expenses payable under the deposit agreement. Any dividend we declare will be distributed by the depositary bank to the holders of the ADSs, subject to the terms of the deposit agreement. See “Description of American Depositary Shares — Dividends and Other Distributions.”
37
The following table sets forth our cash and our capitalization as of December 31, 2020, on:
• an actual basis; and
• an as adjusted basis to give effect to the issuance and sale of ADSs in this offering at the assumed initial public offering price of US$ per ADS, and an ADS-to-ordinary share ratio of 1-to-50, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
You should read this information in conjunction with our consolidated financial statements and the related notes included elsewhere in this prospectus, the information set forth in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and other financial information contained elsewhere in this prospectus.
|
As of December 31, 2020 (Unaudited) |
||||||||||
|
Actual |
As Adjusted(1)(2) |
|||||||||
|
(in A$ or US$, except share data) |
||||||||||
|
Total cash |
A$11,840,308 |
|
US$9,119,405 |
|
A$ |
US$ |
||||
|
Contributed equity: 1,040,136,110 |
A$45,076,484 |
|
US$34,717,908 |
|
A$ |
US$ |
||||
|
Accumulated losses |
(35,407,592 |
) |
(27,270,927 |
) |
||||||
|
Reserves |
2,133,611 |
|
1,643,307 |
|
|
|
||||
|
Total equity |
11,802,503 |
|
9,090,288 |
|
|
|
||||
|
Total capitalization |
A$11,802,503 |
|
US$9,090,288 |
|
A$ |
US$ |
||||
____________
(1) The as adjusted statement of financial position data give effect to our receipt of net proceeds from the issuance and sale of ADSs at the assumed initial offering price of US$ per ADS, and an ADS-to-ordinary share ratio of 1-to-50, after deducting underwriting commissions and estimated offering expenses payable by us.
(2) Each US$1.00 increase (decrease) in the assumed initial public offering price of US$ per ADS, after giving effect to the ADS-to-ordinary share ratio of 1-to-50, would increase (decrease) each of cash, contributed equity, total equity and total capitalization by A$ million (or US$ million), assuming the number of ADSs offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, an increase (decrease) of 1,000,000 ADSs offered by us would increase (decrease) each of cash, contributed equity, total equity and total capitalization by A$ million (or US$ million), assuming the assumed initial public offering price of US$ per ADS, after giving effect to the ADS-to-ordinary share ratio of 1-to-50, remains the same, and after deducting underwriting discounts and commissions. The as adjusted information is illustrative only and will depend on the actual initial public offering price, number of ADSs offered and other terms of this offering determined at pricing.
The outstanding ordinary share information in the table above is based on 1,040,136,110 ordinary shares outstanding as of December 31, 2020, and excludes:
• 334,855,128 ordinary shares issuable upon the exercise of outstanding options as of December 31, 2020 with a weighted-average exercise price of A$0.11 per ordinary share; and
• 23,287,265 ordinary shares issuable upon exercise of performance rights.
38
If you invest in the ADSs in this offering, your ownership interest will be immediately diluted to the extent of the difference between the initial public offering price per ADS and the as adjusted net tangible book value per ordinary share or ADS immediately after this offering.
As of December 31, 2020, our historical net tangible book value was A$11,802,503 (or US$9,090,288), or A$ (or US$ ) per ADS. Historical net tangible book value per ADS represents our total tangible assets less total liabilities, divided by the number of ordinary shares outstanding as of December 31, 2020, converted to ADSs at an ADS-to-ordinary share ratio of 1-to-50.
After giving effect to the receipt of the net proceeds from our sale of ADSs in this offering at an assumed initial public offering price of US$ per ADS, after deducting underwriting discounts and commissions and estimated offering expenses payable by us, our as adjusted net tangible book value as of December 31, 2020 was A$ million (or US$ million), or A$ (or US$ ) per ADS, equivalent to A$ (or US$ ) per ordinary share, in each case based on an ADS-to-ordinary share ratio of 1-to-50. This represents an immediate increase in net tangible book value of A$ (or US$ ) per ADS, equivalent to A$ or (US$ ) per ordinary share, to our existing shareholders and immediate dilution of A$ (or US$ ) per ADS, equivalent to A$ (US$ ) per ordinary share, to investors purchasing ADSs in this offering, in each case based on an ADS-to-ordinary share ratio of 1-to-50.
The following table illustrates this dilution on a per ADS basis, assuming all ordinary shares outstanding as of December 31, 2020 converted to ADSs at an ADS-to-ordinary share ratio of 1-to-50:
|
Assumed initial public offering price per ADS |
US$ |
|||
|
Historical net tangible book value per ADS as of December 31, 2020 |
US$ |
|||
|
Increase in net tangible book value per ADS attributed to investors purchasing ADSs in this offering |
|
|
||
|
As adjusted net tangible book value per ADS after this offering |
||||
|
Dilution in net tangible book value per ADS to investors in this offering |
US$ |
Each US$1.00 increase (decrease) in the assumed initial public offering price of US$ per ADS, after giving effect to the ADS-to-ordinary share ratio of 1-to-50, would increase (decrease) the as adjusted net tangible book value per ADS after this offering by US$ and dilution to investors in this offering by US$ per ADS, assuming that the number of ADSs offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting underwriting discounts and commissions. An increase of 1,000,000 ADSs offered by us would increase the as adjusted net tangible book value by US$ per ADS and the dilution to investors in this offering would decrease by US$ per ADS, assuming the assumed initial public offering price remains the same and after deducting underwriting discounts and commissions. A decrease of 1,000,000 ADSs offered by us would decrease the as adjusted net tangible book value by US$ per ADS and the dilution to investors in this offering would increase by US$ per ADS, assuming the assumed initial public offering price remains the same and after deducting underwriting discounts and commissions.
If the underwriter exercises its option to purchase additional ADSs in full, the as adjusted net tangible book value after the offering would be US$ per ADS, the increase in net tangible book value per ADS to existing shareholders would be US$ per ADS and the dilution per ADS to new investors in this offering would be US$ per ADS, in each case assuming an initial public offering price of US$ per ADS, after giving effect to the ADS-to-ordinary share ratio of 1-to-50.
The dilution information above is for illustration purposes only. Our as adjusted net tangible book value following the closing of this offering will depend on the actual initial public offering price and other terms of this offering determined at pricing.
39
The following table summarizes, as of December 31, 2020:
• the total number of ordinary shares purchased from us by existing shareholders and the equivalent number of ordinary shares underlying ADSs purchased by investors in this offering;
• the total consideration paid to us by our existing shareholders and by investors purchasing ADSs in this offering, assuming an initial public offering price of US$ per ADS, after giving effect to the ADS-to-ordinary share ratio of 1-to-50, before deducting underwriting discounts and commissions and estimated offering expenses payable by us in connection with this offering; and
• the average price per ordinary share paid by existing shareholders and the average price per ADS or equivalent number of ordinary shares.
|
Ordinary Shares |
Total Consideration |
Average |
Average |
|||||||||
|
Number |
Percent |
Amount |
Percent |
|||||||||
|
Existing shareholders |
|
|
US$ |
|
US$ |
US$ |
||||||
|
Purchasers of ADSs |
|
|
|
|
|
|
||||||
|
Total |
|
100 |
US$ |
100 |
US$ |
US$ |
||||||
If the underwriter exercises its option to purchase additional ADSs in full, our existing shareholders would own % and investors in this offering would own % of the total number of ordinary shares outstanding (including shares underlying ADSs) upon the closing of this offering.
Each US$1.00 increase (decrease) in the assumed initial public offering price of US$ per ADS, after giving effect to the ADS-to-ordinary share ratio of 1-to-50, would increase (decrease) the total consideration paid by investors in this offering by US$ million and increase (decrease) the total consideration paid by investors in this offering by %, assuming that the number of ADSs offered by us, as set forth on the cover page of this prospectus, remains the same and before deducting underwriting discounts and commissions.
The outstanding ordinary share information in the table above is based on 1,040,136,110 ordinary shares outstanding as of December 31, 2020, and excludes:
• 334,855,128 ordinary shares issuable upon the exercise of outstanding options as of December 31, 2020 with a weighted-average exercise price of A$0.11 per ordinary share; and
• 23,287,265 ordinary shares issuable upon exercise of performance rights.
To the extent any outstanding options are exercised, there will be further dilution to investors purchasing in this offering.
40
Selected Consolidated Financial Data
The following tables contain selected portions of our consolidated financial and other data. The selected consolidated statement of profit or loss and other comprehensive income data for the six months ended December 31, 2020 and 2019 and consolidated statement of financial position data as of December 31, 2020 have been derived from our unaudited condensed consolidated financial statements included elsewhere in this prospectus. The selected consolidated statement of profit or loss and other comprehensive income data for the years ended June 30, 2020 and 2019 and consolidated statement of financial position data as of June 30, 2020 have been derived from our audited consolidated financial statements included elsewhere in this prospectus. Our audited consolidated financial statements have been prepared in accordance with IFRS, as issued by the IASB, as of and for the years ended June 30, 2020 and 2019.
You should read the consolidated financial and other data set forth below in conjunction with our consolidated financial statements and the accompanying notes and the information in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” contained elsewhere in this prospectus.
Consolidated Statement of Profit or Loss and Other Comprehensive Income Data
|
Six months ended December 31, |
Year Ended June 30, |
|||||||||||
|
2020 |
2019 |
2020 |
2019 |
|||||||||
|
(in A$, except share amounts) |
(in A$, except share amounts) |
|||||||||||
|
Revenue |
1,177,163 |
|
7,350 |
|
604,884 |
|
— |
|
||||
|
Product costs |
(537,939 |
) |
(8,450 |
) |
(450,345 |
) |
— |
|
||||
|
Research and development costs |
(2,039,147 |
) |
(313,426 |
) |
(2,110,639 |
) |
(736,140 |
) |
||||
|
Loss after income tax expense from continuing operations |
(2,889,389 |
) |
(1,925,473 |
) |
(3,929,284 |
) |
(1,426,198 |
) |
||||
|
Net loss |
(2,889,389 |
) |
(2,212,004 |
) |
(4,697,636 |
) |
(2,718,399 |
) |
||||
|
Loss per share from continuing |
(0.32 |
) |
(0.30 |
) |
(0.57 |
) |
(0.32 |
) |
||||
|
Loss per share from continuing operations and discontinued operations – basic and diluted (in A$ cents) |
(0.32 |
) |
(0.34 |
) |
(0.69 |
) |
(0.61 |
) |
||||
|
Weighted average number of ordinary shares outstanding – basic and diluted |
902,054,732 |
|
649,048,889 |
|
684,035,399 |
|
447,439,263 |
|
||||
|
Dividends per share |
— |
|
— |
|
— |
|
— |
|
||||
Consolidated Statement of Financial Position
|
As of |
As of |
|||||
|
(in A$) |
||||||
|
Cash |
11,840,308 |
|
3,603,390 |
|
||
|
Net assets |
11,802,503 |
|
3,164,428 |
|
||
|
Total assets |
12,180,952 |
|
4,236,079 |
|
||
|
Total liabilities |
378,449 |
|
1,071,651 |
|
||
|
Accumulated losses |
(35,407,592 |
) |
(32,518,203 |
) |
||
|
Issued capital |
45,076,484 |
|
34,192,043 |
|
||
41
Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following “Management’s Discussion and Analysis of Financial Condition and Results of Operations” should be read together the section titled “Selected Consolidated Financial Data” and our consolidated financial statements and the accompanying notes included elsewhere in this prospectus. This discussion includes both historical information and forward-looking information based upon current expectations that involve risk, uncertainties and assumptions. Our actual results may differ materially from management’s expectations as a result of various factors, including, but not limited to, those discussed in “Risk Factors” and elsewhere in this prospectus.
Overview
We are a development stage enterprise at an early stage in the development of our drug candidate. We have incurred net losses since inception and expect to incur substantial and increasing losses for the next several years as we expand our research and development activities and move our drug candidate into later stages of development. The process of carrying out the development of our drug candidates to later stages of development may require significant additional research and development expenditures, including pre-clinical testing and clinical trials, as well as for obtaining regulatory approval. To date, we have funded our operations primarily through the sale of equity securities, proceeds from the exercise of options, grants and interest income
The financial statements for fiscal year 2020, 2019 and for the half-year report ended on December 31, 2020 and 2019 are presented without the consolidation of the Company’s wholly-owned subsidiary which was sold on June 30, 2020.
Operating Results
Results of Operations
Comparison of Fiscal Year Ended June 30, 2020 to June 30, 2019
The following tables set forth our results of operations in Australian dollars for the years ended June 30, 2020 and 2019.
|
Year ended June 30, |
||||||
|
2020 |
2019 |
|||||
|
A$ |
A$ |
|||||
|
Revenue |
604,884 |
|
— |
|
||
|
Other income |
217,170 |
|
1,553 |
|
||
|
Product costs |
(450,345 |
) |
— |
|
||
|
Administration expense |
(457,673 |
) |
(330,178 |
) |
||
|
Advertising and promotion |
(406,225 |
) |
(94,814 |
) |
||
|
Research and development costs |
(2,110,639 |
) |
(736,140 |
) |
||
|
Compliance, legal and regulatory |
(235,163 |
) |
(72,181 |
) |
||
|
Finance cost |
— |
|
(85,065 |
) |
||
|
Share based payments |
(565,448 |
) |
(47,854 |
) |
||
|
Occupancy expenses |
(2,085 |
) |
(1,519 |
) |
||
|
Salaries and employee benefit expense |
(523,760 |
) |
(60,000 |
) |
||
|
Loss after tax from continuing operations |
(3,929,284 |
) |
(1,426,198 |
) |
||
|
Loss after tax from discontinuing operations |
(768,352 |
) |
(1,292,201 |
) |
||
|
Total comprehensive loss for the year |
(4,697,636 |
) |
(2,718,399 |
) |
||
Revenue
Revenue increased to A$604,884 in fiscal year 2020 from no revenue in fiscal year 2019, primarily due to the commencement of sales of the cannabinoid oil products.
42
Other Income
Other income increased to A$217,170 in fiscal year 2020 from A$1,553 in fiscal year 2019, primarily due to settlement agreements of terminated contractual arrangements and receipt of government based COVID assistance funding.
Product costs
Production costs increased to A$450,345 in fiscal year 2020 from no production costs in fiscal year 2019, primarily due to the costs involved in the production of the cannabinoid oil products.
Administration expense
Administration expense increased to A$457,673 in fiscal year 2020 from A$330,178 in fiscal year 2019, primarily due to an increase in listing and registry costs which was partially offset by the full repayment in 2019 of the convertible notes outstanding.
Advertising and promotion
Advertising and promotion expense increased to A$406,225 in fiscal year 2020 from A$94,814 in fiscal year 2019, primarily due to the costs of marketing of the cannabinoid oil products.
Research and development costs
Research and development costs increased to A$2,110,639 in fiscal year 2020 from A$736,140 in fiscal year 2019, primarily due an increase in the development of our clinical trials, particularly the acquisition of patient data to identify unmet medical conditions that our cannabinoid oil products were being used to treat. Patient data was acquired through a research service provided by our oil distributor. Research costs in 2020 also increased due to the commencement of TBI and inflammatory disease preclinical studies and the OSA proof of concept clinical trial. We did not track research and development costs by development program or by the nature of the research and development cost in fiscal year 2019 or 2020 but are implementing a system to do so moving forward.
Compliance, legal and regulatory
Compliance, legal and regulatory expense increased to A$235,163 in fiscal year 2020 from A$72,181 in fiscal year 2019, primarily due to the regulatory requirements to conduct clinical trials and also to sell the cannabinoid oil products.
Finance cost
Finance Costs decreased from A$85,065 to nil in the six months ended on December 31, 2020, primarily due to the engagement during the six months ended on December 31, 2019 of consultants to assist in the development of financial and capital raising strategies.
Share based payments
Share-based payments expense increased to A$565,448 in fiscal year 2020 from A$47,854 in fiscal year 2019, primarily due to the costs associated with an increased number of equity issuances.
Occupancy expenses
Occupancy expenses increased to A$2,085 in fiscal year 2020 from A$1,519 in fiscal year 2019, with no significant changes during these years.
Salaries and employee benefit expense
Salaries and employee benefit expense increased to A$523,760 in fiscal year 2020 from A$60,000 in fiscal year 2019, primarily due to an increase in headcount.
43
Loss after tax from continuing operations
Loss after tax from continuing operations increased to A$3,929,284 in fiscal year 2020 from A$1,426,198 in fiscal year 2019, primarily due to an increase in the expenses for the development of our clinical trials and the expenses associated with our share-based payments.
Loss after tax from discontinuing operations
Loss after tax from discontinuing operations decreased to A$768,352 in fiscal year 2020 from A$1,292,201 in fiscal year 2019, primarily due to the sale of the segment relating to the oral device business.
Comparison of Six Months Ended December 31, 2020 to December 31, 2019
The following tables set forth our results of operations in Australian dollars for the six months ended December 31, 2020 and 2019.
|
Six months ended December 31, |
||||||
|
2020 |
2019 |
|||||
|
A$ |
A$ |
|||||
|
Sales |
1,177,163 |
|
7,350 |
|
||
|
Product costs |
(537,939 |
) |
(8,450 |
) |
||
|
Other income |
52,078 |
|
2,929 |
|
||
|
Administration expense |
(454,664 |
) |
(173,228 |
) |
||
|
Advertising and investor relation |
(227,532 |
) |
(141,783 |
) |
||
|
Compliance, legal and regulatory |
(89,065 |
) |
(56,270 |
) |
||
|
Research and development costs |
(2,039,147 |
) |
(313,426 |
) |
||
|
Share based payment expense |
(380,371 |
) |
(966,937 |
) |
||
|
Occupancy expenses |
(61,992 |
) |
(1,042 |
) |
||
|
Salaries and employee benefit expense |
(327,920 |
) |
(274,616 |
) |
||
|
Loss after tax from continuing operations |
(2,889,389 |
) |
(1,925,473 |
) |
||
|
Loss after tax from discontinuing operations |
— |
|
(286,531 |
) |
||
|
Total comprehensive loss for the period |
(2,889,389 |
) |
(2,212,004 |
) |
||
Sales
Sales increased to A$1,177,163 in the six months ended on December 31, 2020 from A$7,350 in the six months ended on December 31, 2019, primarily due to the commencement of sales of the cannabinoid oil products as of December 2019.
Other income
Other income increased to A$52,078 in the six months ended on December 31, 2020 from A$2,929 in the six months ended on December 31, 2019, primarily due to receipt of government based COVID assistance funding.
Product costs
Product costs increased to A$537,939 in the six months ended on December 31, 2020 from A$8,450 in the six months ended on December 31, 2019, primarily due to the costs involved in the production of the cannabinoid oil products.
Administration expense
Administration expense increased to A$454,664 in the six months ended on December 31, 2020 from A$173,228 in the six months ended on December 31, 2019, primarily due to an increase in listing and registry costs, recruitment costs, and consultancy fees.
44
Advertising and investor relation
Advertising and investor relation expense increased to A$227,532 in the six months ended on December 31, 2020 from A$141,783 in the six months ended on December 31, 2019, primarily due to the costs of marketing of the cannabinoid oil products.
Research and development costs
Research and development costs increased to A$2,039,147 in the six months ended on December 31, 2020 from A$313,426 in the six months ended on December 31, 2019, primarily due an increase in the development of our clinical trials, particularly the acquisition of patient data to identify unmet medical conditions that our cannabinoid oil products were being used to treat. Patient data was acquired through a research service provided by our oil distributor. Research costs in 2020 also increased due to the commencement of TBI and inflammatory disease preclinical studies and the OSA proof of concept clinical trial. We did not track research and development costs by development program or by the nature of the research and development cost in the six months ended on December 31, 2019 or 2020 but are implementing a system to do so moving forward.
Compliance, legal and regulatory
Compliance, legal and regulatory expense increased to A$89,065 in the six months ended on December 31, 2020 from A$56,270 in the six months ended on December 31, 2019, primarily due to the regulatory requirements to conduct clinical trials and also to sell the cannabinoid oil products.
Share based payments
Share-based payments expense decreased to A$380,371 in the six months ended on December 31, 2020 from A$966,937 in the six months ended on December 31, 2019, primarily due to the reduction in equity issuances.
Occupancy expenses
Occupancy expenses increased to A$61,992 in the six months ended on December 31, 2020 from A$1,042 in the six months ended on December 31, 2019, primarily due to the company relocating offices and incurring additional expenses during this process.
Salaries and employee benefit expense
Salaries and employee benefit expense increased to A$327,920 in the six months ended on December 31, 2020 from A$274,616 in the six months ended on December 31, 2019, primarily due to an increase in headcount.
Loss after tax from continuing operations
Loss after tax from continuing operations increased to A$2,889,389 in the six months ended on December 31, 2020 from A$1,925,473 in the six months ended on December 31, 2019, primarily due to an increase in the expenses for the development of our clinical trials.
Loss after tax from discontinuing operations
Loss after tax from discontinuing operations decreased to no loss in the six months ended on December 31, 2020 from A$286,531 in the six months ended on December 31, 2019, primarily due to the sale of the segment relating to the oral device business.
Liquidity and Capital Resources
Since our inception, our operations have mainly been financed through the issuance of equity securities. Additional funding has come through interest earned from cash on term deposit.
45
Equity Issuances
The following table summarizes our issuances of ordinary shares for cash, share-based payments and executive and employee compensation in the last two fiscal years.
|
Fiscal |
Number of |
Net |
||||
|
(in A$) |
||||||
|
Ordinary Shares (net of costs) |
2019 |
293,608,792 |
2,184,801 |
|||
|
Ordinary Shares (net of costs) |
2020 |
166,757,449 |
7,469,392 |
Capital Requirements
As of December 31, 2020, we had year-end cash of A$11,840,308. We anticipate that our current cash will be sufficient to fund our operations for more than 12 months from the date of this filing. However, our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement that involves risks and uncertainties, and actual results could vary materially. If we are unable to raise additional capital when required or on acceptable terms, we may have to significantly delay, scale back or discontinue one or more of our clinical trials or our operations.
We anticipate that we will require substantial additional funds in order to achieve our long-term goals and complete the research and development of our current drug candidates. We do not expect to generate significant revenue until we obtain regulatory approval to market and sell our drug candidate and sales of our drug candidate have commenced. We therefore expect to continue to incur substantial losses in the near future.
Our future capital requirements are difficult to forecast and will depend on many factors, including but not limited to:
• the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights;
• the scope, results and timing of preclinical studies and clinical trials;
• the costs and timing of regulatory approvals; and
• the costs of establishing sales, marketing and distribution capabilities.
Cash Flows
Audited Financial Years
Comparison of cash flows for the Year ended June 30, 2020, with June 30, 2019
The following table summarizes our cash flows for the periods presented:
|
Year ended June 30, |
||||||
|
2020 |
2019 |
|||||
|
A$ |
A$ |
|||||
|
Net cash used in operating activities |
(3,907,334 |
) |
(2,160,433 |
) |
||
|
Net cash provided by (used in) investing activities |
13,000 |
|
(22,942 |
) |
||
|
Net cash provided by financing activities |
7,404,392 |
|
2,049,801 |
|
||
Operating Activities
Net cash used in operating activities increased to A$3,907,334 in 2020 from A$2,160,433 in 2019, primarily due to the expansion of our clinical trials.
Investing Activities
Net cash provided by investing activities increased to A$13,000 in 2020 from A$22,942 net cash used in 2019, primarily due to asset sales associated with the discontinuance of the dental devices business.
46
Financing Activities
Net cash provided by financing activities increased to A$7,404,392 in 2020 from A$2,049,801 in 2019, primarily due to the exercise of options and private placements of ordinary shares that raised A$7,469,392 in 2020.
Unaudited Interim Period
Comparison of cash flows for the six months ended December 31, 2020, with December 31, 2019
The following table set forth the sources and uses of cash for the six months ended on December 31:
|
Six months ended December 31, |
||||||
|
2020 |
2019 |
|||||
|
A$ |
A$ |
|||||
|
Net cash used in operating activities |
(2,894,560 |
) |
(1,526,783 |
) |
||
|
Net cash used in investing activities |
29,276 |
|
— |
|
||
|
Net cash provided by financing activities |
11,102,203 |
|
6,561,516 |
|
||
Operating Activities
Net cash used in operating activities increased to A$2,894,560 in the six months ended December 31, 2020 from A$1,526,783 in the six months ended December 31, 2019, due to the expansion of our clinical trials.
Investing Activities
Net cash provided by in investing activities was A$29,276 in 2020 as a result of the sale of Gameday International Pty Ltd.
Financing Activities
Net cash provided by financing activities increased to A$11,102,202 in the six months ended December 31, 2020 from A$6,561,516 in the six months ended December 31, 2019, due to the exercise of options and private placements of ordinary shares.
Critical Accounting Policies and Estimates
The preparation of the consolidated financial statements requires management to make judgements, estimates and assumptions that affect the reported amounts in the financial statements. Management continually evaluates its judgements and estimates in relation to assets, liabilities, contingent liabilities, revenue and expenses. Management bases its judgements, estimates and assumptions on historical experience and on other various factors, including expectations of future events, management believes to be reasonable under the circumstances. The resulting accounting judgements and estimates will seldom equal the related actual results. The judgements, estimates and assumptions that have a significant risk of causing a material adjustment to the carrying amounts of assets and liabilities within the next financial year are discussed here below.
Coronavirus (COVID-19) pandemic
Judgement has been exercised in considering the impacts that the COVID-19 pandemic has had, or may have, on the consolidated entity based on known information. This consideration extends to the nature of the products and services offered, customers, supply chain, staffing and geographic regions in which the consolidated entity operates. Other than as addressed in specific notes, there does not currently appear to be either any significant impact upon the financial statements or any significant uncertainties with respect to events or conditions which may impact the consolidated entity unfavorably as at the reporting date or subsequently as a result of the COVID-19 pandemic.
Share-based payment transactions
The consolidated entity measures the cost of equity-settled transactions with employees and third parties by reference to the fair value of the equity instruments at the date at which they are granted. The fair value is determined by using either the trinomial or Black-Scholes model taking into account the terms and conditions upon
47
which the instruments were granted. The accounting estimates and assumptions relating to equity-settled share-based payments would have no impact on the carrying amounts of assets and liabilities within the next annual reporting period but may impact profit or loss and equity. Refer to notes 13 and 19 of the notes to the financial statements for further information.
Trend Information
We are a clinical stage pharmaceutical development company and it is not possible for us to predict with any degree of accuracy the outcome of our research or commercialization efforts.
Our primary expenditure involves research and development costs. Increases or decreases in research and development expenditure are attributable to the level of clinical trial activity and the amount of expenditure on those trials.
Off-Balance Sheet Arrangements
During fiscal years 2020 and 2019, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
Tabular Disclosure of Contractual Obligations
As of December 31, 2020, our contractual obligations were as set forth below:
|
Payments Due by Period A$ |
||||||||||
|
Total |
Less than |
1 – 3 |
3 – 5 |
More than |
||||||
|
Lease obligations |
95,000 |
56,500 |
38,500 |
— |
— |
|||||
|
Other contractual obligations |
— |
— |
— |
— |
— |
|||||
Contingent liabilities
We did not have any material contingent liabilities outstanding as of December 31, 2020.
Capital commitments
We did not have any material future capital expenditure outstanding as of December 31, 2020.
We have agreements with clinical sites and contract research organizations. We make payments to these sites and organizations based upon the number of patients enrolled and the period of follow-up in the trial.
48
Overview
Our legal name is Incannex Healthcare Limited (“Incannex”). We were incorporated in Australia in April 2001 under the name Mount Magnet South Limited. In November 2016, we changed our name to Impression Healthcare Limited, and in June 2020, to Incannex Healthcare Limited. Incannex is listed on the ASX under the symbol “IHL.”
Strategy
Our mission is to create premier ethical pharmaceutical drugs and therapies for patients with unmet medical needs, in all instances fulfilling regulatory requirements of the Food and Drug Administration (“FDA”) and other relevant regulatory agencies (EMEA, TGA). We aim to be recognized as a leading specialty drug development company, committed to restoring health and transforming the lives of patients through the development of novel pharmaceutical products and treatments.
We develop targeted and scientifically validated fixed-dose combinations of synthetic cannabinoids and psychedelic agents, applying proprietary insights in an effort to create long term value for our patients and shareholders. We focus on clinical indications that we believe represent unmet or inadequately addressed medical needs and also represent compelling commercial opportunities. In particular, we are developing three unique pharmaceutical compositions to target five indications: obstructive sleep apnea (“OSA”), traumatic brain injury (“TBI”)/concussion, rheumatoid arthritis, inflammatory bowel disease and inflammatory lung conditions (“ARDS”, “COPD”, Asthma, Bronchitis). We are also developing a treatment for generalized anxiety disorder (“GAD”) utilising psilocybin combined with innovative psychotherapy methods. We are pursuing FDA registration and marketing approval for each product and therapy under development.
Additionally, we seek to secure patents on our drug candidates in conjunction with our medical and scientific staff, advisors and the investigators of our research studies that constitute our advisory board. Our advisory board is comprised of industry and academic experts familiar with our business, and we meet with the advisory board regularly. The current members of our advisory board are Dr. Sud Agarwal (our Chief Medical Officer and Director), Mark Bleakley (our Head of Programs), Rosemarie Walsh (our Clinical Research Manager), Terrance O’Brien (principal investigator of the IHL-42X from Alfred Hospital), Dr Jennifer Walsh (professor at University of Western Australia), Ron Jithoo (neurosurgeon and advisor for IHL-216), and Paul Liknaitsky (psychedelic principal investigator from Monash University). Our advisory board also comprises our collaborative partners, and in particular Monash University, The Alfred Hospital and the University of Western Australia Centre for Sleep Science.
To achieve our goals, we intend to:
• Advance our novel investigational drug candidates towards approval in the United States and elsewhere. We are pursuing FDA approval of all our drug candidates currently in development. All preclinical and clinical trials are structured to ensure that each program is FDA compliant. We will be pursuing a New Drug Application (“NDA”) with the FDA with respect to each of our drug candidates. If the NDA is approved, the product may be marketed in the United States. Once an NDA for one of our drug candidates is approved in the United States, we plan to pursue marketing approval of our drug candidates in other regions including the Europe Union, Japan, Australia and Israel.
• Take advantage of accelerated commercialization pathway options for our drug candidates. We and our regulatory consultants believe that each of our drug candidates will qualify for one or more FDA expedited review programs (breakthrough designation, accelerated approval, priority review and/or fast track), as there are a limited amount of pharmaceutical drug treatments approved in the U.S. to treat the indications that we are targeting with our drug candidates, and the pharmaceutical treatments that do exist provide limited treatment and are costly. These expedited review programs
49
often result in accelerated and less-costly regulatory pathways to approval compared with traditional regulatory pathways. We have not yet approached the FDA about the suitability of our products for these accelerated approval pathways and such designations do not guarantee accelerated review by the FDA.
• Develop future clinical products targeting unmet medical needs. We intend to only develop clinical products that treat unmet medical conditions. As a result, we may have opportunities to accelerate commercialization of such products.
• Maintain a strong intellectual property portfolio. We have developed a global intellectual property strategy to support our commercial objectives. We are monitoring the results of our research and development programs to identify new intellectual property that aligns with those commercial objectives. We intend to take a global approach to our intellectual property strategy and we intend to pursue patent protection in key global markets, including the United States, Europe, Japan and Israel. We have pending patent applications relating to our drug candidates IHL-42X, IHL-216A and IHL-675A. This approach aligns with our regulatory strategy, including the proposed submission of Pre-Investigational New Drug Application (“pre-IND”) meeting requests to the FDA for our clinical programs.
Clinical Approach
We are pursuing FDA approval of all our drug candidates currently being developed. We will be working with the FDA to ensure each clinical program is structured to meet regulatory requirements. FDA approval will be sought following the completion of successful phase 3 studies. Once we receive FDA approval for our drug candidates, we will be able to commercialize our drug candidates in the United States and pursue regulatory approval for the drug to be made available in other jurisdictions, including the Europe, Japan, Australia and Israel. The graphic below represents our clinical development pipelines.
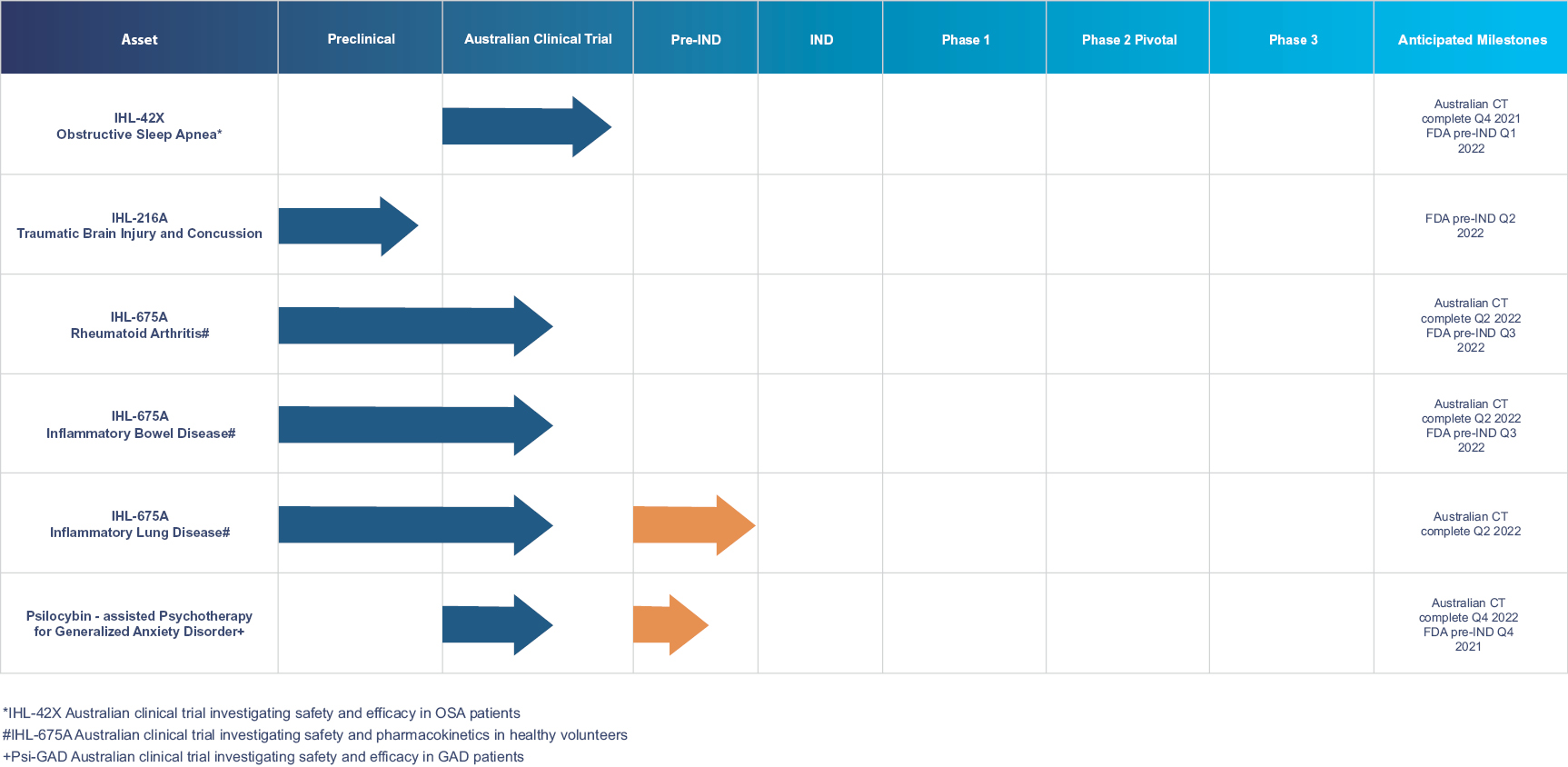
Market Opportunity
The combined annual global market size of the indications we are targeting is over US$110 billion, which is derived from the total addressable market for the treatment of OSA, TBI, concussions, rheumatoid arthritis, inflammatory bowel disease, inflammatory lung conditions (ARDS, COPD, Asthma, Bronchitis) and GAD. Thus, there is significant economic potential to shareholders, as well as benefit to patients suffering from these medical conditions.
50
Our Drug Candidates
IHL-42X
Obstructive Sleep Apnea
Obstructive sleep apnea is characterized by a narrowing or obstruction of the upper airway in sleep, interfering with breathing and interrupting sleep. This relatively common and chronic disorder is underdiagnosed and inadequately treated. It is understood to contribute to a wide range of serious long-term outcomes, including cardiovascular disease, cognitive impairments such as memory loss, poor concentration and judgment, depression and death or injury due to traffic accidents resulting from excessive daytime sleepiness. The costs associated with OSA are substantial, relating to lost productivity, workplace and motor vehicle accidents.
A 2019 article published by the Lancet premised on literature-based analysis of 17 studies across 16 countries, estimated that OSA affects some 936 million adults worldwide. This alarming statistic is also thought to be increasing due to growing prevalence of obesity and an ageing global population. Many people with OSA develop high blood pressure (hypertension), which can increase the risk of cardiovascular disease. The more severe the OSA, the greater the risk of coronary artery disease, heart attack, heart failure and stroke.
There are no registered drugs for OSA. Current treatment options include: continuous positive airway pressure (“CPAP”) in which an external device pneumatically splints the airway open to prevent disruptions in breathing; oral appliances to advance the mandible or to retain the tongue, putting the mouth in a position more conducive to breathing; surgery to remove physical obstructions to air flow; and implantable electronic stimulators to activate muscles at the base of the tongue, opening the airway in synchrony with respiration. However, all of these therapies are inadequate, expensive, and for implantable stimulators and surgery, invasive.
The standard treatment option is the mechanical CPAP device, however, we believe patient compliance to CPAP devices is low due to discomfort and claustrophobia resulting from pressurized air being pumped into the patient’s nose and/or mouth during sleep. Despite these discomforts, the global annual market for OSA detection and treatment using CPAP devices is over US$10 billion and growing.
IHL-42X in Obstructive Sleep Apnea
IHL-42X is a fixed-dose combination of acetazolamide, a registered pharmaceutical, and dronabinol, a synthetic form of -Delta-9-tetrahydrocannabinol (THC); both agents have been shown to reduce the apnea hypopnea index (“AHI”). We believe that the activity of dronabinol on cannabinoid receptors causes dilation of the airway, and acetazolamide induces modest metabolic acidosis, signalling to the body that there is excess CO2 in the blood, thus increasing respiration. By exploiting two mechanisms that both reduce AHI in one pharmaceutical formulation, we believe that IHL-42X can have a therapeutic benefit at doses of each constituent drug that are safe and tolerable.
Australian Stage 2 Clinical Trial for IHL-42X for Obstructive Sleep Apnea (“OSA”)
We are currently conducting a proof-of-concept Phase 2 clinical trial in Australia to support our IND application with the FDA and to inform the clinical design of our future pivotal Phase 2 clinical trial, which will be conducted under the IND to assess the safety and efficacy of IHL-42X in patients with Obstructive Sleep Apnea. The IND for IHL-42X in treatment of OSA has not yet been submitted and although we have incorporated multiple facets into this study, including full monitoring by a CRO and CDISC data formatting, there is no guarantee that the FDA will accept data from the Australian trial and further testing may be required prior to opening the NDA.
We received approval from The Alfred Hospital Human Research Ethics Committee in September 2020 to proceed with the trial in Australia. In December 2020, we recruited the first patients to the randomized, double-blind, placebo-controlled clinical trial that assesses the therapeutic benefit of IHL-42X at three different doses. The primary endpoint of the trial is the change in AHI relative to baseline and the secondary endpoints are change in oxygen desaturation index (“ODI”), daytime somnolence measured by the Epworth Sleepiness Scale, improvement in mood as measured by the POMS (Profile of Moods State), and well-being as measured by the Short Form 36 and the safety of the IHL-42X combination will be established through adverse event monitoring.
51
The study is currently underway and well-advanced at the Alfred Hospital in Melbourne Australia and the University of Western Australia Centre for Sleep Science in Perth. We have retained Novotech, a global contract research organization, to manage and to monitor the study. In July 2021, an interim analysis of the data from our ongoing phase 2b double blind randomized placebo-controlled clinical trial was performed and these results have been utilized to support a patent application regarding the methods for the treatment of obstructive sleep apnea. Additionally, we plan to supply IHL-42X for sale in Australia under the Special Access Scheme, a system in Australia that allows certain health practitioners to access therapeutic goods, such as unregistered medicinal cannabinoid products, that are not included in the Australian Register of Therapeutic Goods, after the completion of the pivotal Phase 2 study and prior to drug registration.
IHL-216A
IHL-216A for Concussion/Traumatic Brain Injury and Chronic traumatic encephalopathy
Concussion/Traumatic Brain Injury are caused by a rapid acceleration/deceleration of the brain caused by a direct blow to the head or sudden impact to the body that jolts the skull. This causes the brain to compress against the skull. The impact of the brain against the skull causes both macro and micro scale damage to the brain which sets of a series of physiological events called secondary injury cascades. These secondary injury cascades are what cause many of the neurocognitive deficits seen in TBI patients.
Falls, vehicle collisions, violence, sports and combat injuries are the main activities leading to TBI and concussion. The signs and symptoms of a concussion can be subtle and may not show up immediately. Symptoms can last for days, weeks or even longer. Common symptoms after a concussive traumatic brain injury are headache, loss of memory (amnesia) and confusion. The amnesia usually involves forgetting the event that caused the concussion. Other symptoms include nausea, vomiting, fatigue, blurry vision and ringing in the ears.
Complications can occur immediately or soon after a traumatic brain injury. Severe injuries increase the risk of a greater number of and more-severe complications. Moderate to severe traumatic brain injury can result in prolonged or permanent changes in a person’s state of consciousness, awareness or responsiveness. Many people who have had a significant brain injury will experience changes in their cognitive ability, have executive functioning problems and or communication, emotional and behavioral problems. Some research suggests that repeated or severe traumatic brain injuries might increase the risk of degenerative brain diseases, but this risk cannot be predicted for an individual.
Chronic traumatic encephalopathy (“CTE”) is the term used to describe brain degeneration likely caused by repeated head traumas. CTE is a diagnosis made only at autopsy by studying sections of the brain. CTE is a rare disorder that is not yet well understood. CTE is not related to the immediate consequences of a late-life episode of head trauma. CTE has a complex relationship with head traumas such as persistent post-concussive symptoms and second impact syndrome that occur earlier in life.
Experts are still trying to understand how repeated head traumas, including how many head injuries and the severity of those injuries, and other factors might contribute to the changes in the brain that result in CTE.
CTE has been found in the brains of football players, boxers and other athletes that play contact sports, along with military personnel who were exposed to explosive blasts. Some signs and symptoms of CTE are thought to include difficulties with thinking (cognition) and emotions, physical problems and other behaviors. Symptoms of CTE often manifest decades after head trauma occurs.
CTE cannot be made as a diagnosis during life except in those rare individuals with high-risk exposures. Researchers do not yet know the frequency of CTE in the population and do not understand the causes. There is no cure for CTE. Researchers are currently developing diagnostic biomarkers for CTE, but none have been validated yet.
52
IHL-216A Formulation development for clinical trials
IHL-216A is a fixed dose combination of isoflurane, a registered pharmaceutical, and CBD, intended for administration in the immediate period after primary blunt head injury to prevent development of brain injuries. Isoflurane is approved in the United States for induction and maintenance of anaesthesia. CBD is approved for use in seizure disorders and has shown effects on neuroinflammatory responses to brain injury. Isoflurane is a registered pharmaceutical, and also has demonstrated neuroprotective activity (neuroprotective activity, or neuroprotection, is defined as reduced neuronal cell death or disruption) in animal studies of TBI and is thought to act by modulating glutamate release and calcium uptake as well as via effects on mitochondrial membrane depolarization and excitatory neurotransmission. Thus, we believe that IHL-216A may affect neuroexcitation, neuro-inflammation, cerebral blood flow and cerebral oxygen consumption resulting in overall neuroprotection. We are also assessing its ability to protect the brain against secondary injury mechanisms that cause neuronal cell death and raised intracranial pressure in the days and weeks following head trauma in sports, and all other applicable scenarios resulting in head trauma (falls, vehicle collisions, violence, combat, among other causes). Reducing secondary brain injury may improve positive outcomes for long term neurological sequelae, including CTE, a major health risk associated with contact sports.
The formulation of IHL-216A presents unique challenges. Because isoflurane is an inhaled volatile anesthetic, it cannot be used in a typical oral drug combination product. We intend to formulate IHL-216A as a combined inhalational product. Nebulized drug delivery involves using air pressure or ultrasonic vibrations to turn a liquid drug solution into an aerosol. We engaged Vectura, a UK based contract development and manufacturing organization, to develop the nebulised CBD formulation and device for delivery of the CBD to the isoflurane anaesthetic circuit. Development of the nebulized CBD formulation will be an iterative process starting with three steps of refinement based on properties of the solution, generated aerosol and dose delivery. Vectura specializes in the development of inhaled drugs and has an excellent track record of bringing products to market and have formulated pharmaceutical drugs for multinational pharmaceutical companies including Bayer, Sandoz and Novartis.
Appointing Vectura to develop the IHL-216A formulation in parallel with the animal study using the NFL model of concussion will ensure that we are readied with the specific formulation and delivery mechanism required for advancement of a pivotal Phase 2 clinical trial once the Stage 2 in vivo study and formulation is finalized.
Due to the product’s potential therapeutic utility in contact sports, IHL216A is being designed to satisfy the World Anti-doping Authority (“WADA”) specifications for use by athletes at risk of TBI and CTE.
Stage 1 pre-clinical study for IHL-216A for TBI and CTE
In December 2020, we completed an animal study to formally assess the neuroprotective capability of IHL-216A. The study introduced rodents to head trauma in a highly controlled manner to inflict a reproducible injury. Various doses of IHL-216A or its active pharmaceutical ingredients were administered to eight cohorts of rodents soon after traumatic head injury. Behavioral tests were used to assess the neurocognitive and motor function over time. We also monitored secondary injury cascades, and performed micro-scale cellular analysis post-mortem to discern and compare neuronal damage across the cohorts.
As detailed below, we found that the IHL-216A components, CBD and isoflurane, act synergistically to reduce indicators of neuronal damage, neuroinflammation and behavioral deficits that are consequences of TBI, as IHL-216A outperformed the predicted effect of CBD and isoflurane combined. The predicted result is determined by analyzing the results of isoflurane and CBD independently, and then based on those results predicting how well the drugs would do in combination; to the extent IHL-216A exceeds the predicted result, we can conclude that the drugs strengthen the effectiveness of one another and synergy exists. The study also found that IHL-216A reduced neuronal damage, neuroinflammation and cognitive deficits in a rodent model of TBI to a greater extent than either CBD or isoflurane applied on a standalone basis. These results have not been assessed for statistical significance.
Post-mortem analysis of rat brains also detected synergy between CBD and isoflurane. Brains were fixed and sectioned prior to Nissl staining to identify neuronal damage. Nissl staining is a microscopy technique to visualise Nissl bodies. Healthy neurons typically have more Nissl bodies than damaged ones. Neuronal damage is indicated by the ratio of Nissl bodies to neurons across different sections of the hippocampus with a lower Nissl/neuron ratio indicative of increased neuronal damage. Synergy between CBD and isoflurane was detected in hippocampal regions cornu ammonis 1 (CA1) and cornu ammonis 2 (CA2). These regions of the brain are known to be important in the
53
formation and storage of memories. In the study, the improvement in Nissl/Neuron ratio observed for IHL-216A treated animals was increased by 53% for CA1 and 60% for CA2 relative to CBD alone, 28% for CA1 and 145% for CA2 relative to isoflurane alone, and by 20% for CA1 and 53% for CA2 relative to the predicted effect of CBD and isoflurane combined. These results demonstrated that less neuronal damage was observed in the rats treated with IHL-216A relative to the predicted value.
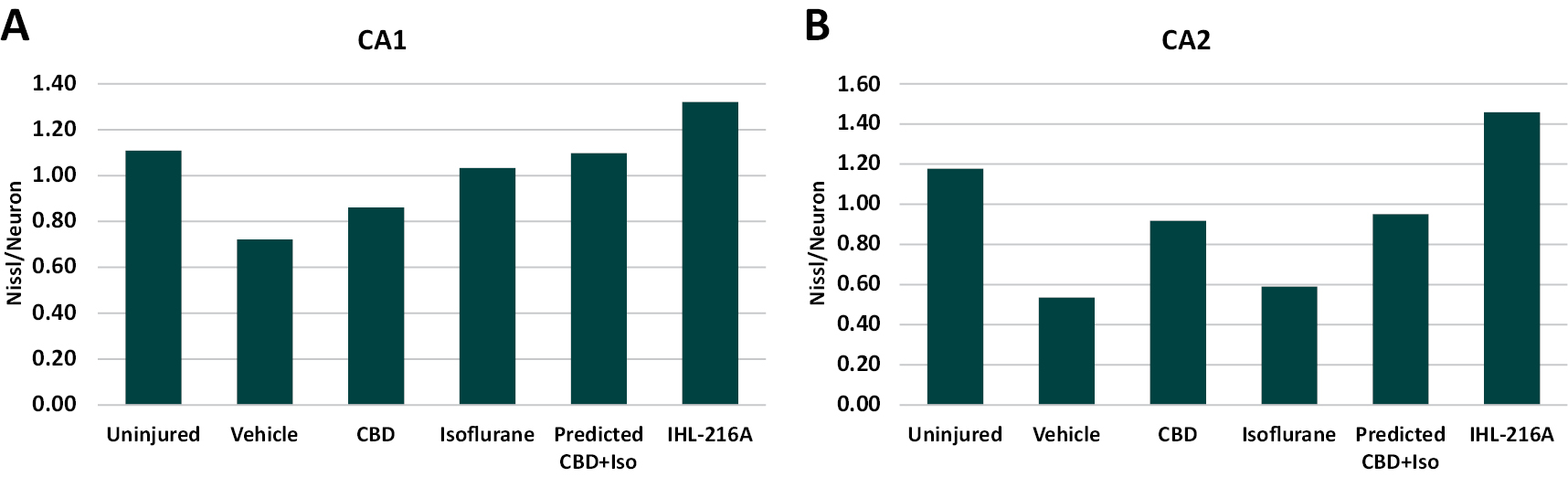
Figure 1. Synergistic activity of CBD and isoflurane (IHL-216A) in neuronal damage as assessed by Nissl staining. Rodents that had undergone TBI and treated with CBD and/or isoflurane or vehicle were assessed for neuronal damage by post-mortem analysis of fixed brain sections by Nissl staining. Nissl staining permits the quantitation of the ratio of Nissl bodies to total neurons, a lower ratio being indicative of increased neuronal damage. The Nissl/neuron ratio observed in hippocampal regions (A) CA1 and (B) CA2 contralateral to the site of injury in the group treated with IHL-216A was greater than that predicted based on the groups treated with each drug alone, which is indicative of synergy. Values are average of the respective treatment group. Group sizes: Uninjured n=6, vehicle n=6, CBD n=6, isoflurane n=5, IHL-216A n=6. Neuroinflammation Marker — Iba1.
A post-mortem analysis of the rat brains also determined that CBD and isoflurane were synergistic in reducing levels of the neuroinflammation marker Iba1 as detected using immunofluorescence. Iba1 is a protein expressed in microglia, a type of innate immune cell in the brain, that is an established marker of microglial activation and neuroinflammation. The levels of Iba1 in the brain are detected using immunofluorescence, which is a microscopy technique that employs antibodies specific to Iba1 which are detected using a fluorescent tag. Increased levels of Iba1 are indicative of increased neuroinflammation. IHL-216A reduced the Iba1 neuroinflammation marker by 35% more than CBD alone and 123% more than isoflurane administered alone. IHL-216A also reduced the Iba1 neuroinflammation marker by 10% more than the predicted value of the combined CBD and isoflurane treatments.
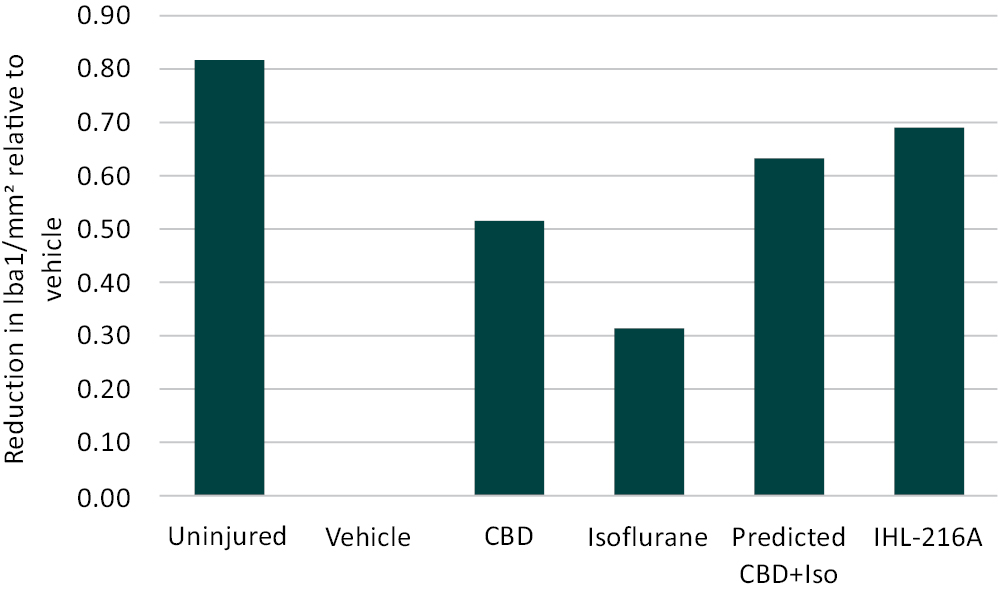
Figure 2. Synergistic activity of CBD and isoflurane (IHL-216A) in reducing levels of the neuroinflammatory marker Iba1. Rodents that had undergone TBI and treated with CBD and/or isoflurane or vehicle were assessed for neuroinflammation through immunofluorescence analysis of the neuroinflammatory marker Iba1. Iba1 levels increase after TBI and a reduction in Iba1 is indicative of a reduction in neuroinflammation. Iba1 levels in brain sections ipsilateral to the site of injury in the group treated with IHL-216A were reduced more than would be predicted based on the reduction observed in groups treated with each drug alone, which is indicative of synergy. Values are average of the respective treatment group. Group sizes: Uninjured n=6, vehicle n=5, CBD n=6, isoflurane n=3, IHL-216A n=5.
54
Synergy between CBD and isoflurane was detected in the behavioral outcomes assessed using the Morris Water Maze. In the Morris Water Maze animals are trained to find a platform in a pool of water. After a number of training sessions, the platform is removed and the mice are monitored to determine whether they return to the location of the platform, which is a measure of spatial learning and memory. Treatment with IHL-216A led to an improvement in the number of times rats returned to the location of the platform per group as well as the proportion of rats in the group that returned to the location of the platform relative to the predicted effect of CBD and isoflurane of 87 % and 24 % respectively. The improved performance of IHL-216A treated rats compared to the predicted effect demonstrated the synergistic effect of CBD and isoflurane.
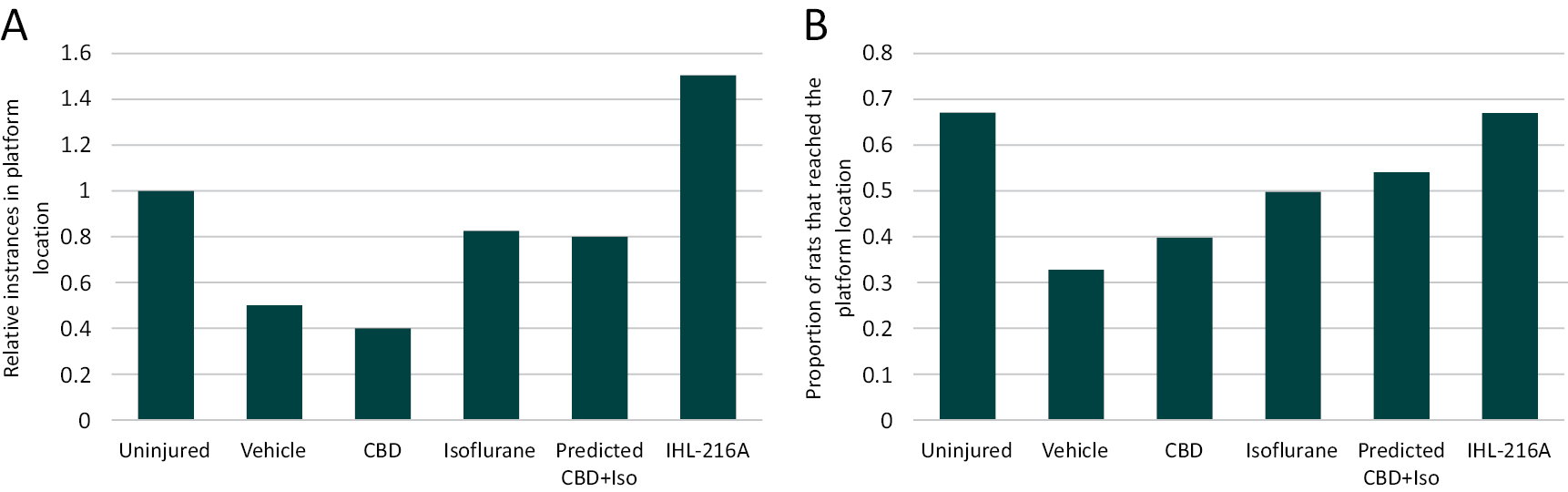
Figure 3. Synergistic activity of CBD and isoflurane (IHL-216A) in the Morris Water Maze assessment. Rodents that had undergone TBI and treated with CBD and/or isoflurane or vehicle were assessed for spatial learning and memory using the Morris Water Maze. The observed performance with respect to both (A) relative instances of animal in platform location and (B) proportion of animals in that reached the platform location was better in the group treated with the CBD isoflurane combination (IHL-216A) than what was predicted based on the performance of the groups treated with each drug alone. This outperformance by the IHL-216A compared to the predicted performance is indicative of synergy. Values are average of the respective treatment group. Group sizes: Uninjured n=6, vehicle n=6, CBD n=5, isoflurane n=6, IHL-216A n=6.
Stage 2 pre-clinical study for IHL-216A
We are currently undertaking a second and more-extensive animal study on the protective effect of IHL-216A in sports concussion with the Monash Trauma Group at the Department of Neuroscience, Monash University, Australia.
The Monash Trauma Group consists of a team of leading scientists within their respective fields. Their research focuses on the effects, underlying pathophysiological mechanisms, biomarkers, and treatments of trauma related conditions including TBI and concussion as well as other types of neurological diseases, including CTE.
The study is coordinated by Dr Stuart McDonald, an expert in fluid biomarker development for monitoring TBI, Associate Professor Richelle Mychasiuk, an expert in animal models of TBI and their clinical relevance, and Associate Professor Sandy Shultz, an expert in the pathological mechanisms, biomarkers and treatments of TBI and related conditions.
The model of TBI being used in this study was developed by Monash University in collaboration with the US National Football League (“NFL”). The results of the study will be used as a precursory data set to inform the pivotal clinical trials required for drug registration. Assessments in this study will include neurocognitive performance, levels of blood biomarkers associated with traumatic brain injury, and post mortem analysis of brain tissue using both MRI and immunohistochemistry.
IHL-675A
IHL-675A comprises a combination of hydroxychloroquine, a registered pharmaceutical, and CBD. Hydroxychloroquine (HCQ) is a disease modifying anti-rheumatic drug that regulates the activity of the immune system, which may be overactive in some conditions. HCQ can modify the underlying disease process, rather than simply treating the symptoms. We have demonstrated that IHL-675A components, cannabidiol and hydroxychloroquine, act synergistically to inhibit production of key inflammatory cytokines in an in vitro study and in 4 distinct successful in vivo experiments using established models of inflammation. We are able to
55
determine whether synergies exist in IHL-675A studies by comparing the predicted result of CBD and HCQ acting together to the actual IHL-675A results. The predicted result is determined by analyzing the results of HCQ and CBD independently in the study, and then based on those results predicting how well the drugs would do on a combined basis; to the extent IHL-675A exceeds the predicted result, we can conclude that the drugs strengthen the effectiveness of one another and synergies exist.
We have evaluated the results of these experiments and believe IHL-675A to be a multi-use candidate for the prevention and treatment of inflammatory lung conditions (ARDS, COPD, asthma, and bronchitis), rheumatoid arthritis and inflammatory bowel diseases. Potentially, this could mean that IHL-675A is a better alternative to CBD based products for certain inflammatory diseases, subject to further examination.
We have completed a pre-IND meeting with the FDA to discuss the regulatory pathway for the development of IHL-675A for lung inflammation in the United States and plan to open INDs for each of the three indications. FDA agreed that marketing applications for IHL-675A should be 505(b)(2) applications due to the existence of certain safety and efficacy information on the active ingredients of IHL-675A originating from historical studies that we are entitled to use in a new drug application. In the context of the IHL-675A development program, this means that we do not have to perform many of the nonclinical toxicology studies that are required for approval of a new chemical entity because there is adequate toxicology data for both CBD and HCQ available in the literature or in regulatory submissions for the respective reference listed drugs. However, we still need to demonstrate IHL-675A is safe and effective in the target indication via a series of randomized, controlled clinical trials.
Lung Inflammation (COPD, Asthma, ARDS and Bronchitis)
Chronic obstructive pulmonary disease (“COPD”) is a chronic inflammatory lung disease that causes obstructed airflow from the lungs. Symptoms include breathing difficulty, cough, mucus (sputum) production and wheezing. It is typically caused by long-term exposure to irritating gases or particulate matter, most often from cigarette smoke. People with COPD are at increased risk of developing heart disease, lung cancer and a variety of other conditions.
Asthma is a condition in which inflammation causes the airways to narrow and swell and which may cause the patient to produce extra mucus. This can make breathing difficult and trigger coughing, a whistling sound (wheezing) during breathing and shortness of breath. For some people, asthma is a minor nuisance. For others, it can be a major problem that interferes with daily activities and may lead to a life-threatening asthma attack. According to Allied Market Research, the Global COPD and asthma drug market is expected to reach US$50.4 billion by 2022, growing at a CAGR of 3.7% from 2016 to 2022.
Acute respiratory distress syndrome (“ARDS”) occurs when fluid builds up in the air sacs (alveoli) located in the lungs. The fluid prevents oxygen from reaching the bloodstream. This deprives organs of the oxygen they need to function. ARDS typically occurs in people who are already critically ill or who have significant injuries. Severe shortness of breath (the main symptom of ARDS) usually develops within a few hours to a few days after the primary injury or infection. It is the one of the main causes of death resulting from COVID-19 and many people who develop ARDS do not survive. The risk of death increases with age and severity of illness. People who survive ARDS may experience lasting damage to their lungs.
Bronchitis is an inflammation of the lining of the bronchial tubes of the lungs. Bronchitis may be either acute or chronic. While acute bronchitis is common, chronic bronchitis, a more serious condition, is a constant irritation or inflammation of the lining of the bronchial tubes.
Rheumatoid Arthritis
Rheumatoid arthritis is a chronic inflammatory disorder that can affect joints, skin, eyes, lungs, heart and blood vessels. As an autoimmune disorder, rheumatoid arthritis is caused by attacks to body tissues by one’s immune system. Unlike the wear-and-tear damage caused by osteoarthritis, rheumatoid arthritis causes a painful swelling that can eventually result in bone erosion and joint deformity.
HCQ is approved for treatment of rheumatoid arthritis in the form of hydroxychloroquine sulphate and marketed as Plaquenil. HCQ has risks of ocular toxicity and cardiac effects including cardiomyopathy and QT prolongation amongst long term users, as listed in the prescribing material.
56
Similarly, long term use of HCQ in rheumatoid arthritis patients was associated with increased cardiovascular mortality. Therefore, there is value in reducing the dose of HCQ in these arthritis patients. To understand the capacity for the combination of CBD with HCQ to permit reduction of the HCQ dose, in an animal study, low dose IHL-675A (1 mg/kg CBD + 2.5 mg/kg HCQ) was compared to a standard dose of HCQ (25 mg/kg HCQ). The 25 mg/kg HCQ dose in rats is equivalent to a 243 mg HCQ dose in a 60 kg human based on the FDA body surface area dose equivalence of 6/37.
In a rheumatoid arthritis animal disease model study, low dose IHL-675A reduced disease severity scores across multiple assessments including clinical score, paw volume, pannus score, total histology score and serum cytokine levels to a greater extent than the equivalent of a standard dose of HCQ. The reduction in disease severity scores by low dose IHL-675A was 1.06-3.52 times that observed for HCQ alone at the standard dose equivalent.
This indicates that the combination of CBD and HCQ in IHL-675A has the potential to permit a ten-fold reduction in HCQ dose, when combined with CBD, without sacrificing efficacy in treatment of arthritis.
We have broadened claims within initial patent filings to cover rheumatoid arthritis as an indication. We are continuously monitoring the results of our research and development program, with a view to identifying and protecting new IP that aligns with our commercial objectives.
Inflammatory Bowel Disease
Inflammatory Bowel Disease (“IBD”) is an umbrella term used to describe disorders that involve chronic inflammation of the digestive tract. Significant types of IBD include:
• Ulcerative colitis. This condition involves inflammation and sores (ulcers) along the superficial lining of the large intestine (colon) and rectum.
• Crohn’s disease. This type of IBD is characterized by inflammation of the lining of the digestive tract, which often can involve the deeper layers of the digestive tract.
Both ulcerative colitis and Crohn’s disease are usually characterized by diarrhea, rectal bleeding, abdominal pain, fatigue and weight loss. IBD can be debilitating and sometimes leads to life-threatening complications.
The precise cause of inflammatory bowel disease remains unknown. Previously, diet and stress were suspected. However, currently medical practitioners acknowledge that these factors may aggravate, but are not the cause, of IBD. One possible cause is an immune system malfunction. When the immune system attempts to defeat an invading virus or bacterium, an abnormal immune response can cause the immune system to attack the cells in the digestive tract.
Preclinical in vitro study of IHL-675A against inflammation
On November 5, 2020, we released the results of our first in vitro study to investigate the synergistic activity of IHL-675A to inhibit inflammation. To test the anti-inflammatory potential of IHL-675A, human peripheral blood mononuclear cells (“PBMCs”) were stimulated with bacterial lipopolysaccharide (“LPS”). PBMCs were incubated with a range of concentrations of CBD and HCQ in combination or each drug alone and then stimulated with LPS to induce an inflammatory response. The inflammatory response was assessed by measuring cytokine levels in the culture medium after 24 hours. A reduction in cytokine levels in response to drug treatment is indicative of anti-inflammatory activity.
Cytokine levels were averaged across three replicates from two donors and normalized to maximum values to yield a relative inhibition value. A relative inhibition of 1 is complete inhibition of cytokine release whereas a value of 0 is no inhibition of cytokine release. Anti-inflammatory synergy was determined using the standard scientific “Excess over Bliss” (“EOB”) method where the predicted inhibition, as calculated using the formula Epred A+B=(EA+EB)-(EAEB), is subtracted from the observed inhibition to yield an EOB score. An EOB score of greater than zero indicates that the combination is synergistic. None of the below data has been analysed for statistical significance.
57
The study demonstrated that CBD and HCQ act synergistically to inhibit production of the assessed inflammatory cytokines IL-1β, IL-6, TNF-α, IL-1aα, and MIP-1α by PBMCs from the donors. The average EOB scores ranged from 0.32-0.57. The reduction in levels of the five cytokines observed for IHL-675A was 436% to 1320% greater relative to HCQ alone, 109% to 767% greater relative to CBD alone and 87% to 767% greater relative to the predicted combinatorial effect of CBD and HCQ. The results in Figures A, B, C, D and E presented below, display the optimal fixed dose IHL-675A combination assessed for each cytokine. The bars noted as Predicted CBD+HCQ represent what our expectation was based on the activity of each drug individually. The observed inhibition of cytokine release upon treatment with the CBD HCQ combination was greater than predicted based on the activity of each drug alone for each cytokine analyzed.
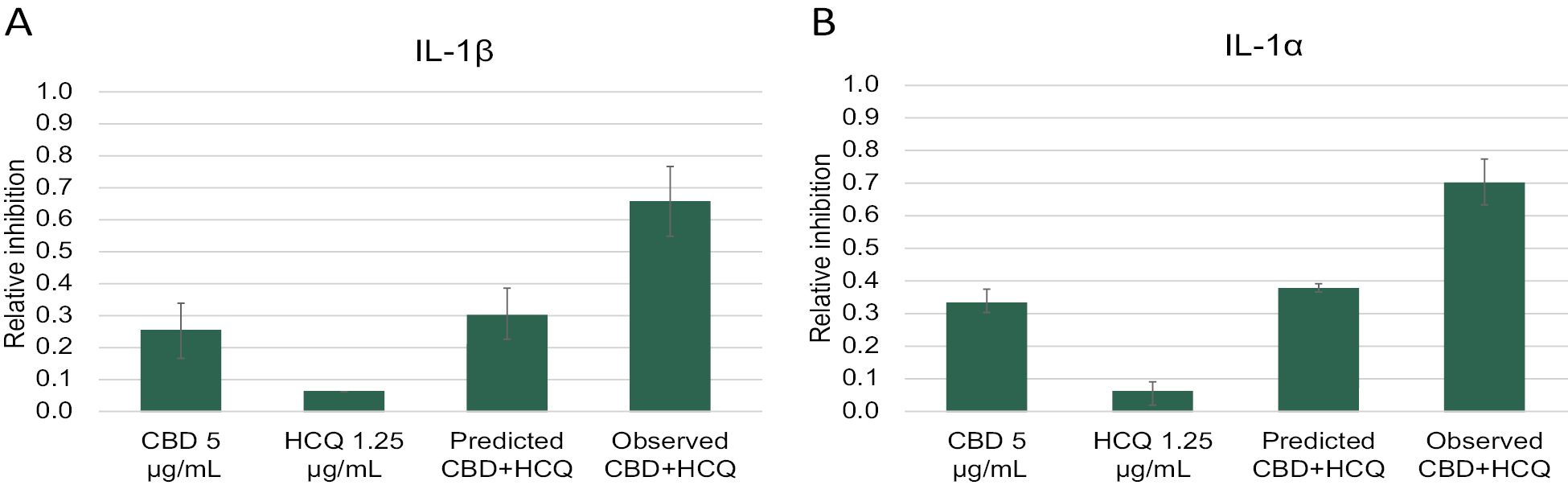
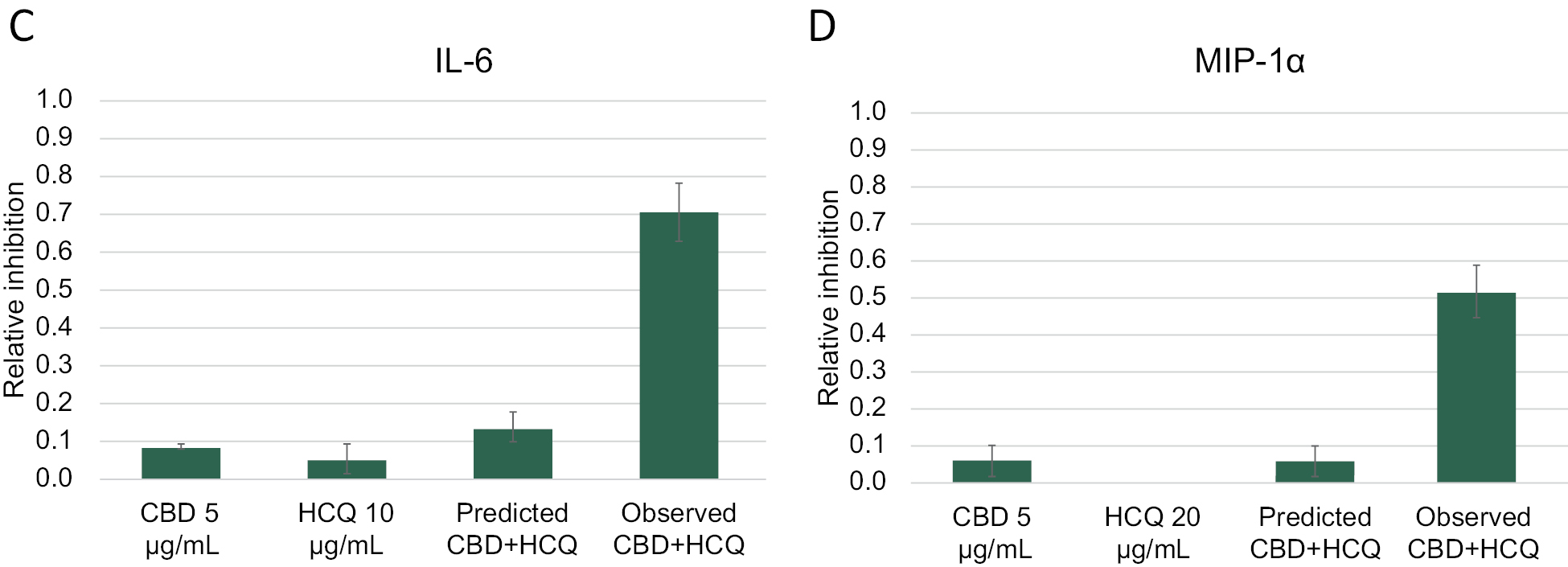
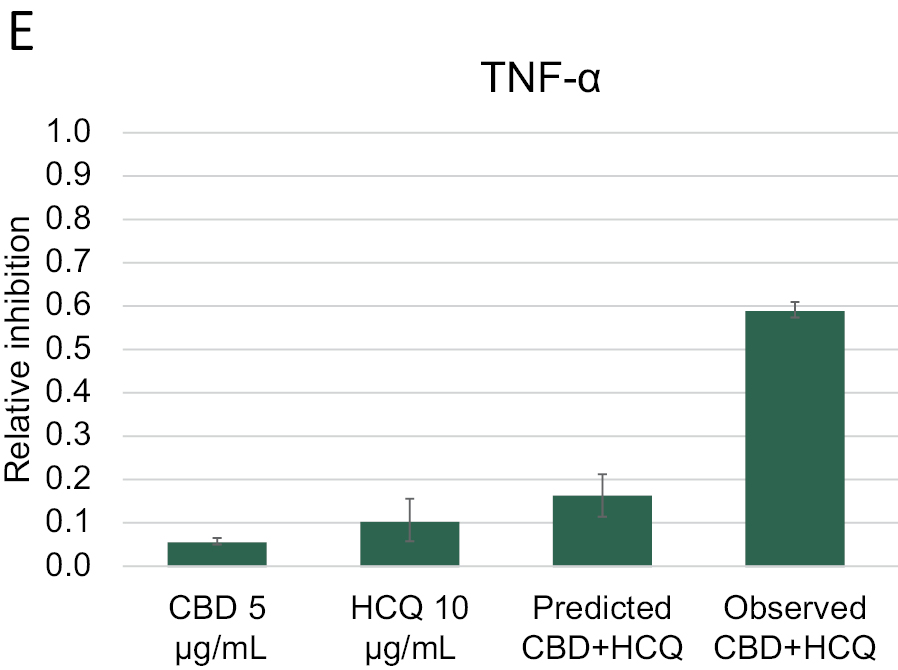
Figure 4. Inhibition of LPS-induced cytokine release from human PBMCs by CBD and HCQ. Data is presented is the average relative inhibition for the PBMC donors. Predicted inhibition by CBD+HCQ was calculated using the formula Epred A+B=(EA+EB)-(EAEB). Observed CBD+HCQ is the level of inhibition observed in the experiment. (A) IL-1b, (B) IL-1a, (C) IL-6, (D) MIP-1a, and (E) TNF-a. Error bars are standard error of the mean of the donors.
58
Preclinical in vivo study of IHL-675A against inflammation
In November of 2020, we announced the results of an in vivo study assessing IHL-675A in a mouse model of sepsis. To determine whether CBD and HCQ synergize in vivo, mice from 11 groups of 10 mice, weighing 18-20g were injected with CBD and HCQ both alone and in combination. After one hour, the mice were injected with LPS to induce an inflammatory response. Each mouse in every cohort was assessed for each of the 5 inflammatory cytokines. Two hours after LPS injection, blood was collected from the mice by cardiac puncture. Sera were processed and analyzed for cytokine levels using a Luminex based assay. For synergy analysis, data was baseline subtracted using sham treated (no LPS injection) cytokine levels and then the values for each cytokine were normalized relative to maximum values across the groups. The normalized values were used to calculate the relative inhibition where a value of 1 is complete inhibition and a value of 0 is no inhibition. Synergy was calculated using the EOB method, or the difference between the observed and predicted inhibition between the combination of drug concentrations where the predicted inhibition is determined using the equation Epred A+B=(EA+EB)-(EAEB). An EOB score of greater than 0 is indicative of synergy.
The results of the in vivo study are presented in Figure 5, showing the optimal fixed dose IHL-675A combination assessed for each cytokine in 11 groups of 10 mice. The bars noted as ‘Predicted CBD + HCQ’ represent IHL’s expectation based on the activity of each drug alone. The observed results from the study significantly exceeded the predicted results across the inflammatory cytokines analyzed. CBD and HCQ synergize to inhibit the production of inflammatory cytokines IL-1β, IL-6, TNF- α, IL12(p70), and IFN-γ in a mouse model of LPS induced sepsis. The average EOB scores ranged from 0.15-0.30. IHL-675A reduced levels of the five inflammatory cytokines to a greater extent than CBD alone. Inhibition of cytokine release by IHL-675A was 26% to 81% greater relative to the predicted effect of the CBD HCQ combination across the five analyzed cytokines after 2 hours.
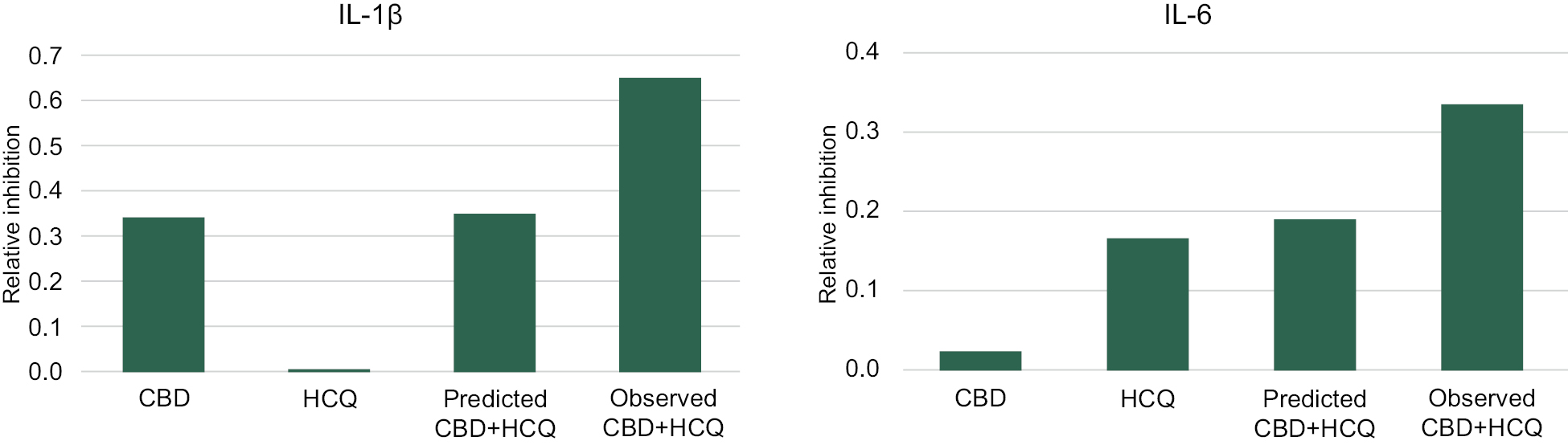
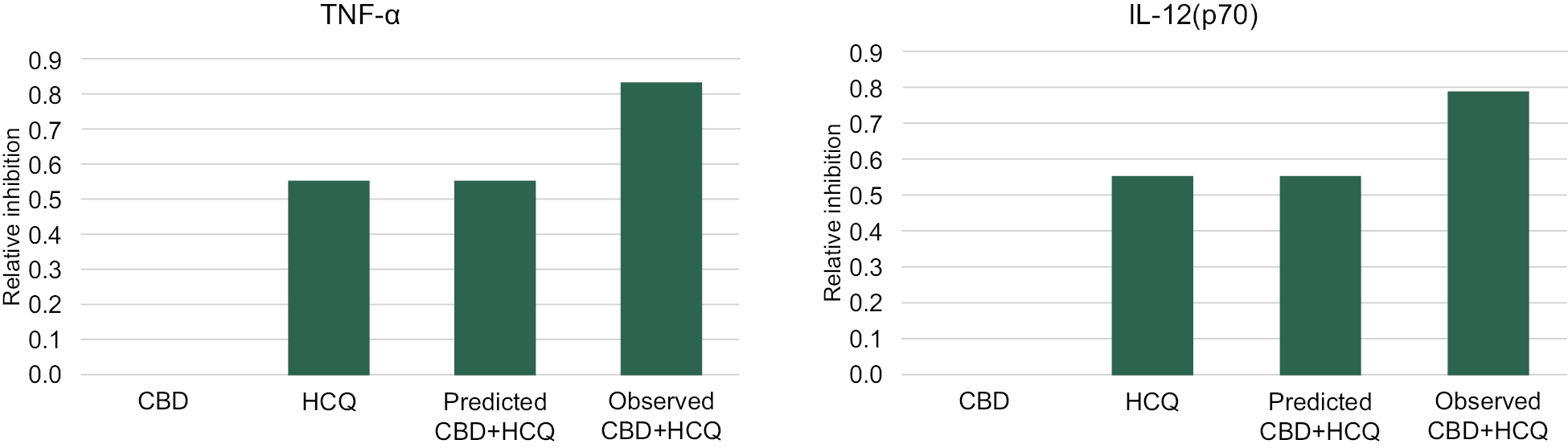
59
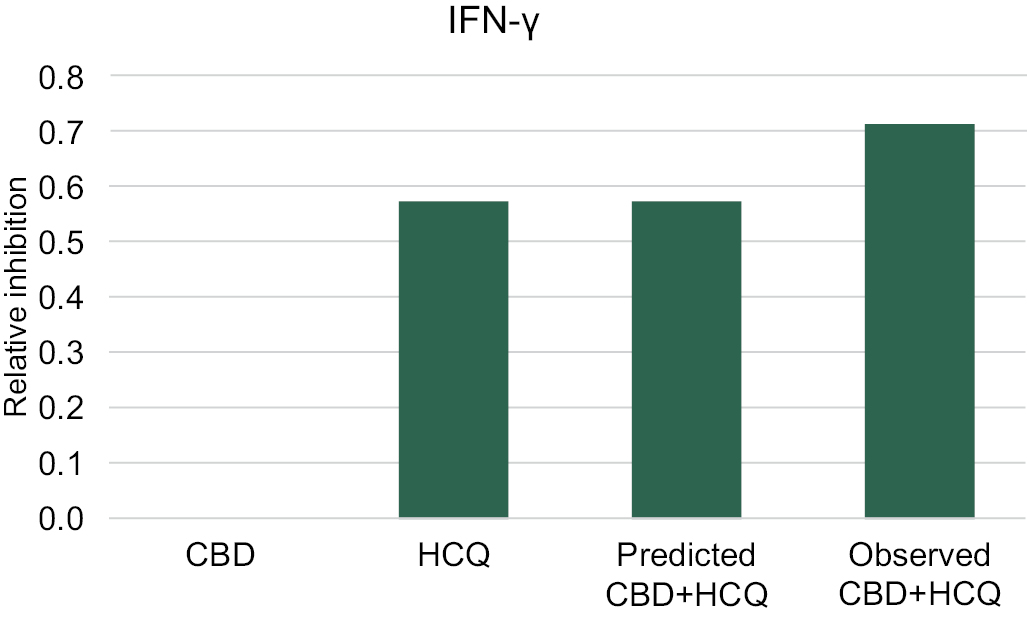
Figure 5. Synergistic anti-inflammatory activity of CBD and HCQ in a mouse sepsis model. The anti-inflammatory activity of the combination of CBD and HCQ was greater than that predicted using the Excess over Bliss method. The CBD+HCQ combination was synergistic at inhibiting release of IL-1β, IL-6, TNF-α, IL12(p70), and IFN-γ.
Preclinical in vivo study of IHL-675A against Pulmonary Inflammation (ARDS, COPD, Asthma and Bronchitis)
In February 2021, we announced the results of an in vivo study assessing IHL-675A anti-inflammatory capabilities regarding chronic obstructive pulmonary disease, asthma, bronchitis, and other inflammatory respiratory conditions. We also assessed the anti-inflammatory effect of our proprietary IHL-675A formulation on Pulmonary Neutrophilia, which is a primary underlying cause of COPD, asthma, bronchitis, and other inflammatory respiratory conditions. We reported encouraging results, as discussed below, which facilitate a substantial expansion of the potential uses for IHL-675A and represent new patient treatment opportunities.
A rodent model of pulmonary inflammation was used to assess the anti-inflammatory efficacy of IHL-675A in lungs. In this study, ten groups of six mice each were pre-treated with either CBD, HCQ or IHL-675A prior to intratracheal administration of bacterial lipopolysaccharide (“LPS”), which was then inhaled and acts as an inflammatory stimulus in the lungs. A sham group where LPS was not administered to the mice was also included as a control. The lungs were flushed with a saline solution 24 hours after LPS administration and bronchoalveolar lavage fluid (‘BALF’) was analyzed for cytokine levels using a Luminex based assay. Cytokines are proteins that mediate the inflammatory response and a reduction in cytokine levels is indicative of reduced inflammation. A white blood cell (‘WBC’) count was also performed on the BALF. When inflammation occurs in the lungs, WBCs are recruited as part of the inflammatory response. A reduction in WBC count is also indicative of reduced inflammation.
60
Cytokine levels were normalized to those detected in vehicle treated mice and then the relative inhibition was calculated. IHL-675A reduced levels of all assessed inflammatory cytokines IL-1β, IL-6, TNF-α, CXCL1 and MCP-1 to a greater extent than either CBD or HCQ alone. WBC counts were normalized using the same method used for cytokines and IHL-675A reduced WBC counts to a greater extent than CBD or HCQ alone. These results indicate that IHL-675A has superior anti-inflammatory activity compared to CBD and HCQ in a mouse pulmonary inflammation model. Based on these results IHL-675A will be assessed for efficacy in the treatment of pulmonary inflammation in humans. These results have not been analysed for statistical significance.
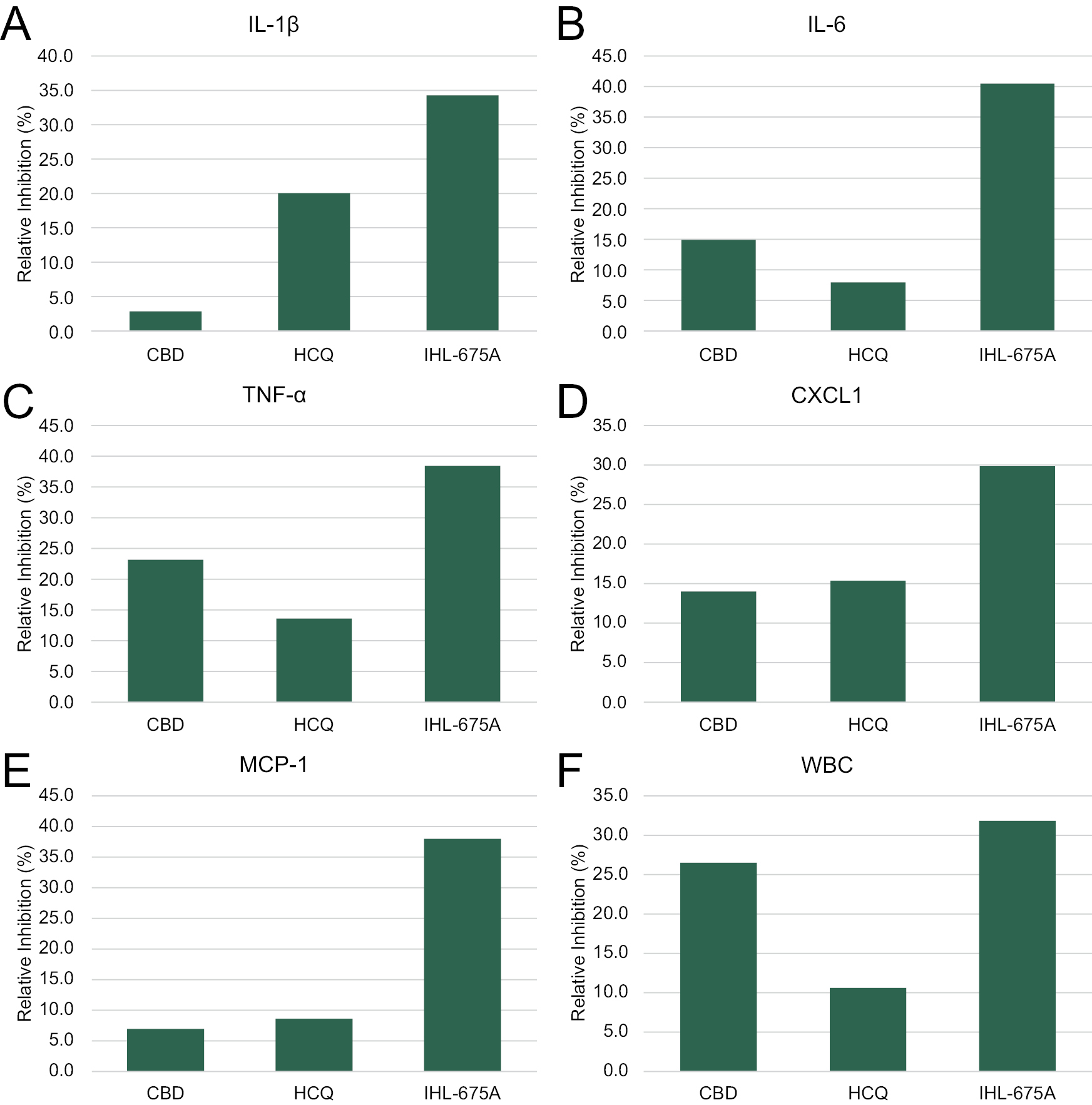
Figure 6. Reduction in cytokine levels and white blood cell count in BALF resulting from treatment with by IHL-675A, CBD or HCQ in a mouse model of pulmonary inflammation. Mice were treated with CBD, HCQ or a combination of CBD and HCQ (IHL-675A) and then LPS was administered intratracheally. Twenty-four hours after LPS administration bronchioalveolar lavage fluid (BALF) was analyzed for cytokine levels and white blood cell count. The reduction in cytokine levels by IHL-675A was greater than that for either drug alone. Drug concentrations were 1 mg/kg CBD and 25 mg/kg HCQ for (A) IL-1β, (B) IL-6, (C) MCP1 and (E) TNF-α, 10 mg/kg CBD and 2.5 mg/kg HCQ for CXCL-1 and WBC (white blood cell count).
61
Preclinical study of IHL-675A in a model of Rheumatoid Arthritis
In March 2021, we announced the results of an in vivo study assessing IHL-675A’s anti-inflammatory capabilities in a rheumatoid arthritis model. Results indicate that a low dose of IHL-675A was 1.06 to 3.52 times more effective at reducing disease severity scores across multiple assessments including clinical score, paw volume, pannus score, total histology score and serum cytokine levels compared to a standard dose of HCQ only. HCQ is approved and widely used for the treatment of rheumatoid arthritis in the form of hydroxychloroquine sulfate, which is marketed as Plaquenil.
In this model of rheumatoid arthritis, female Lewis rats were challenged with porcine type-II collagen with Freund’s adjuvant on Day 1 (0.2 mg/0.2 mL/rat) by subcutaneous injection at the base of the tail to induce arthritis. A booster injection at 0.1 mg/0.1 mL/rat was injected on day 7. On day 16, rats were allocated into groups of six. There were ten groups of modelled rats and one sham injected group. CBD, HCQ or IHL-675A were injected intraperitoneally once per day from day 17 to 30 (total of 14 days). Drug doses were 1 and 10 mg/kg CBD and 2.5 and 25 mg/kg HCQ. The 10 mg/kg CBD and 25 mg/kg HCQ doses were selected as they are representative of standard doses in humans based on the FDA body surface area dose equivalence estimation for rats to humans of 6/37. For a 60 kg person, the 10 mg/kg CBD dose in rats is equivalent to 97 mg and the 25 mg/kg HCQ dose in rats is equivalent to 243 mg. The maintenance dose range recommended for rheumatoid arthritis in the Plaquenil prescribing information is 200-400 mg daily.
Disease severity was assessed by measuring hind paw volume with a plethysmometer and using a qualitative severity score system on days 1, 7 ,10, 14, 16, 18, 20, 22, 24, 26, 28 and 30. Post termination on day 30, blood was collected from all rats and analyzed for levels of the inflammatory cytokines IL-1β and IL-6 using commercially available ELISA kits. These two cytokines were selected as they are known to be involved in the pathophysiology of rheumatoid arthritis. Both hind paws were harvested, weighed and formalin-fixed for histopathology. Histopathological evaluation consisted of an evaluation of cartilage and bone destruction by pannus formation (an abnormal layer of fibrovascular or granulated tissue) and mononuclear cell infiltration in synovial joint tissues. A total histology score, which is a sum of the pannus formation and mononuclear cell infiltration scores, was also calculated. For all assessments, the score was sham subtracted and then the reduction relative to the vehicle group was calculated.
IHL-675A outperformed HCQ alone and CBD alone in the study (at equivalent doses) at reducing clinical score and paw volume at days 24 and 30, pannus formation, total histology score, IL-1β and IL-6 in the rat model of arthritis. The reduction in disease assessments by IHL-675A was 1.07-8.72 times that observed for HCQ alone at an equivalent dose, which indicates that IHL-675A has a benefit in a rat model of arthritis greater than that of HCQ alone and demonstrates that IHL-675A has potential as a treatment for rheumatoid arthritis in humans.
62
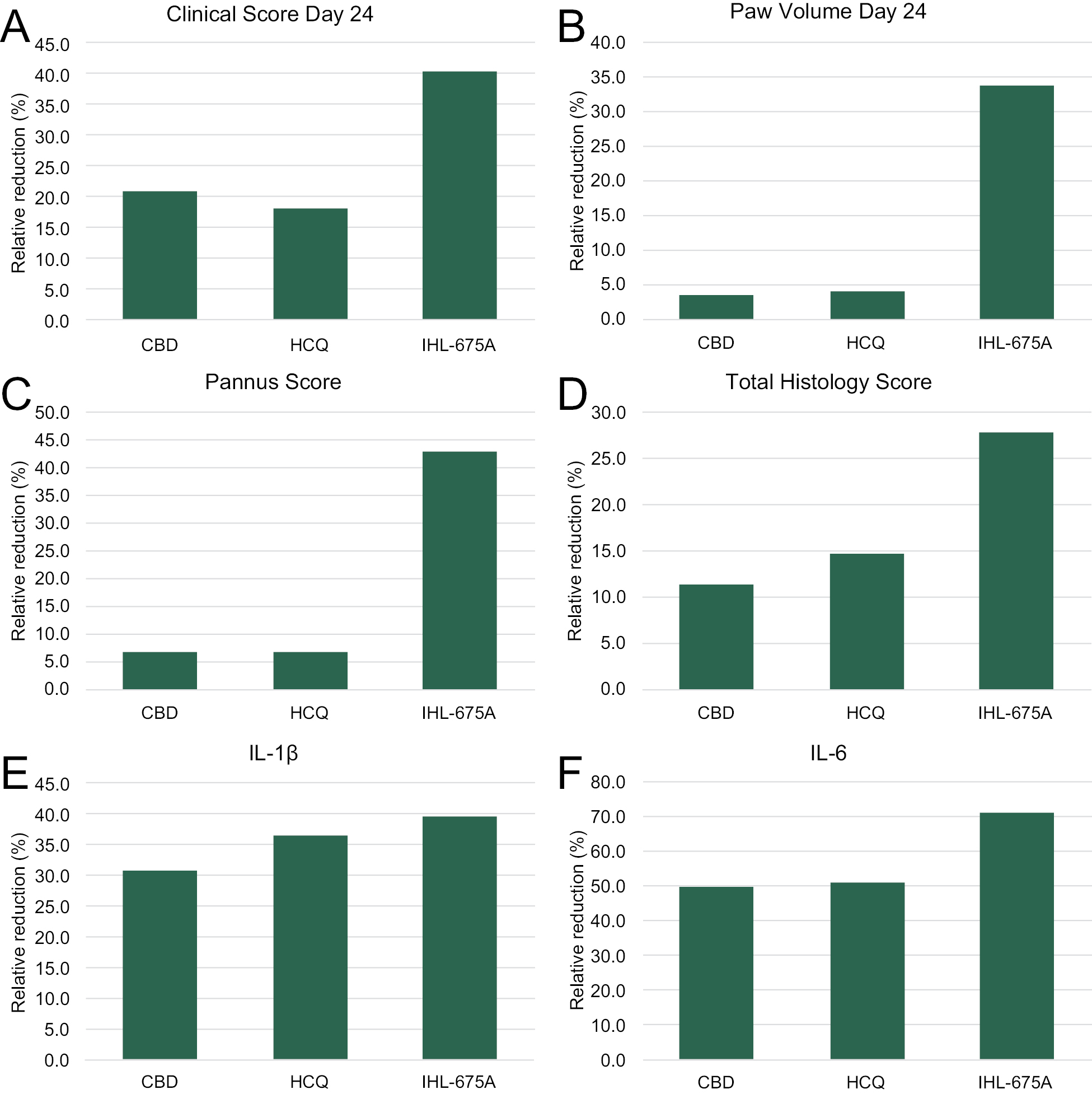
Figure 7. Comparison of IHL-675A to its component drugs CBD and HCQ in reduction of disease assessments in a rat model of rheumatoid arthritis. Groups of rats that had undergone collagen-induced arthritis modelling were treated with IHL-675A, CBD or HCQ at equivalent doses (1 mg/kg CBD, 2.5 mg/kg HCQ). The reduction in arthritis disease severity in IHL-675A treated rats was greater than for either CBD or HCQ treated rats with respect to (A) clinical score at day 24, (B) paw volume at day 24, (C) pannus formation, (D) total histology score, (E) serum IL-1b levels and (F) serum IL-6 levels.
Preclinical studies of IHL-675A in models of inflammatory bowel disease
In February 2021, we announced the results of an in vivo study assessing IHL-675A’s anti-inflammatory capabilities regarding inflammatory bowel disease. IHL-675A demonstrated a reduction in the Colitis index of 46%, while CBD only and HCQ only treatment achieved a reduction of 25% and 27% respectively, demonstrating that IHL-675A has superior anti-inflammatory activity compared to CBD only and HCQ only, which indicates that IHL-675A has the potential to be a treatment for inflammatory bowel disease in humans.
63
This study used eleven groups of six mice. Mice were treated with IHL-675A, CBD or HCQ for four consecutive days after administration of TNBS/ethanol to induce ulcerative colitis. A vehicle treated group and sham group were included in the study. Stool consistency was monitored over the course of the experiment. On Day 5 mice were sacrificed, blood collected for cytokine analysis and the colon removed for analysis.
Endpoint measurements include stool consistency score (an ordinal scale that measures stool consistency with a higher number indicative of looser stools) , colon weight, colon macroscopic damage score (an ordinal scale that combines adhesions, strictures, ulcers/inflammations and instances of wall thickening), colitis index (a composite scale from the histological examination of colon sections) and myeloperoxidase (an enzyme abundantly expressed in neutrophil granulocytes that contributes to inflammatory damage in IBD) levels in the colon tissue at day 5. The results from each of these endpoints were sham subtracted and the relative reduction was calculated. The data was not analysed for statistical significance.
IHL-675A outperformed both CBD alone and HCQ alone at reducing the colitis index, macroscopic damage score, stool consistency score, colon to body weight ratio and myeloperoxidase (MPO) levels. These results indicate that IHL-675A has a benefit in a mouse model of ulcerative colitis greater than that of CBD or HCQ alone, which indicates that IHL-675A is a potential treatment for inflammatory bowel disease in humans.
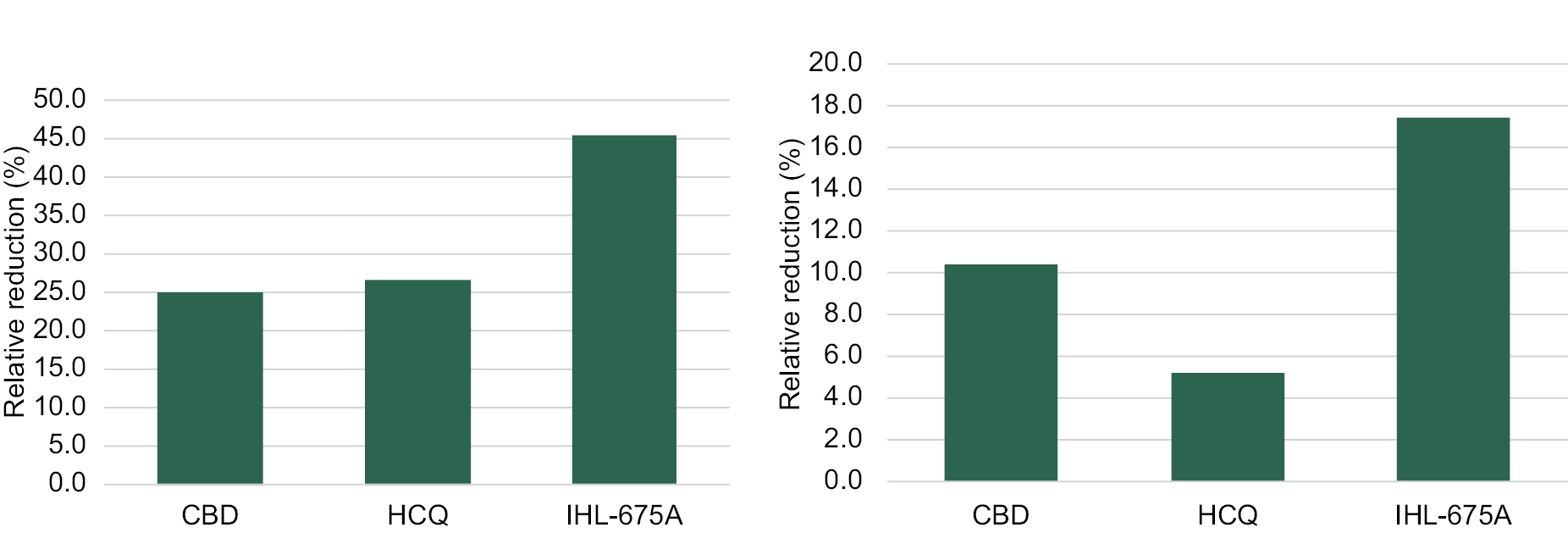
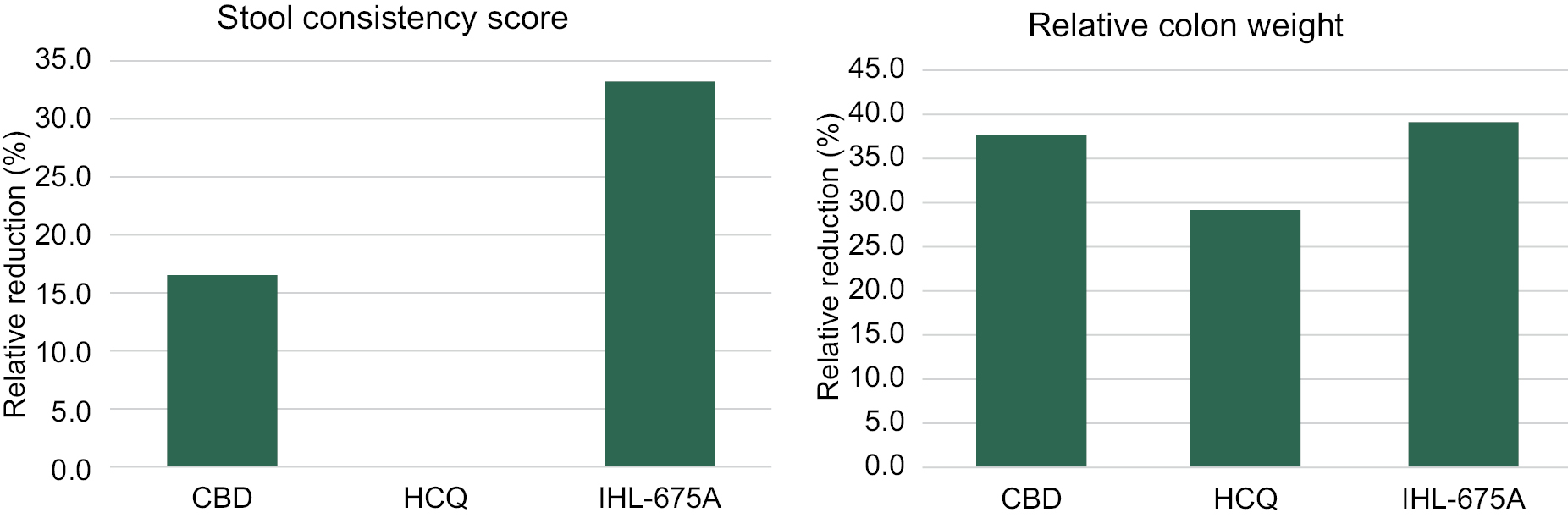
Figure 8. Reduction in colitis score assessments by CBD and HCQ (IHL-675A) in a mouse model of colitis. Colitis was induced in mice by intracolonic installation of TNBS/ethanol and then treated with CBD, HCQ or CBD and HCQ (IHL-675A). After 4 days, mice were sacrifice and the colons extracted for macro and microscopic analysis. The reduction in colitis severity was greater in mice treated with IHL-675A than for either CBD or HCQ alone for (A) colitis index, (B) macroscopic damage score, (C) relative colon weight, (D) stool consistency and (E) MPO levels. Drug dose in all assessments was 1 mg/kg CBD and 2.5 mg/kg HCQ.
Planned phase 1 clinical trial for IHL-675A
We have designed a Phase 1 clinical trial in Australia to assess the safety and pharmacokinetics of IHL-675A in healthy volunteers, the results of which will form part of our FDA IND submissions across the indications of lung inflammation, rheumatoid arthritis and inflammatory bowel disease. The aims of this study are to demonstrate that there are no, or minimal, additional risks/side effects associated with the combination of CBD and HCQ compared
64
to each drug alone and that the uptake and metabolism (pharmacokinetics) of the two drugs do not interfere with one another. A total of 36 subjects will participate in the trial, evenly divided across three arms. The three arms of 12 subjects each will receive one of IHL-675A, Epidiolex (CBD), or Plaquenil (HCQ). The safety and pharmacokinetic assessments will be identical across the three arms.
CBD and HCQ both have both been used historically as treatments for our targeted indications when used independently. However, as with any pharmaceuticals there are risks involved. Part of the strategy in the design of IHL-675A is that the combination of CBD with HCQ permits a reduction in HCQ, which reduces the known risks associated with cumulative HCQ dose, without sacrificing efficacy. Results from the preclinical studies we have conducted to-date have led to the hypothesis that a lower cumulative dose of HCQ, when combined with CBD, will be effective in treating IHL-675A’s targeted indications. Nonetheless, there is always potential for two drugs to interact and exacerbate minor concerns that exist when used alone or lead to new safety concerns. Demonstrating that a combination drug containing CBD and HCQ has a similar safety profile to the component drugs is an important step in the development program and is a requirement set out by regulatory agencies. This clinical trial will be performed in a Phase 1 unit with around the clock monitoring in the event that an adverse event needs to be managed. Safety assessments will include cardiac monitoring via ECG and blood biomarkers, serum liver enzyme levels, blood cell counts and biochemistry, monitoring of vital signs and mental health questionnaires. Due to the substantial evidence of synergy between HCQ and CBD required to produce a superior outcome on inflammatory markers, dosages of HCQ and CBD may be significantly lower than for treatment with the individual drugs and this will be further evaluated in clinical trials.
The other component of this study is monitoring the pharmacokinetics of the two active pharmaceutical ingredients (“API”) of IHL-675A, CBD and HCQ, and comparing them to their respective reference listed drugs Epidiolex and Plaquenil. Study participants will be dosed with either IHL-675A, Epidiolex or Plaquenil with equivalent amounts of the respective API. Blood samples will be drawn at predetermined intervals over a 72-hour period and analyzed for levels of CBD and HCQ as well as their major metabolites. For each molecule the maximum concentration (“Cmax”), time to maximum concentration (“Tmax ”) and total exposure (“AUC”) will be determined. The pharmacokinetic parameters for IHL-675A, Epidolex and Plaquenil will be compared to determine whether the APIs in IHL-675A are bioequivalent to the reference listed drugs. Bioequivalence is an important component of the FDA 505(b)2 approval pathway that IHL is targeting with IHL-675A.
Results from this study will form a component of future regulatory applications for IHL-675A and will also inform the design of Phase 2 efficacy and safety studies across indications.
Psilocybin-assisted Psychotherapy for General Anxiety Disorder (Psi-GAD)
Generalized Anxiety Disorder
Generalized Anxiety Disorder (GAD) is characterized by diffuse, excessive, uncontrollable anxiety that frequently occurs and is not restricted to any particular environmental circumstances. Symptoms are variable, including feelings of persistent and excessive worry, nervousness, restlessness, difficulty in concentrating fatigue, irregular sleeping patterns, muscle tension, irritability, and nausea.
Generalized anxiety disorder is a relatively common and serious psychiatric condition affecting around 4-6% of the population during their lifetime. GAD can severely affect quality of life and professional career prospects.
Existing treatments
International guidelines for GAD treatment recommend selective serotonin reuptake inhibitors (“SSRIs”), serotonin and noradrenaline reuptake inhibitors (“SNRIs”), and pregabalin as first-line options, with benzodiazepines such as diazepam as second-line options. GAD is also treated with psychotherapy alone or in combination with pharmacotherapies. However, these treatments show limited efficacy, with less than half of patients achieving remission following these treatments and substantial treatment side-effects and cost. In particular, the side effects associated with long term use of these pharmacotherapies include emotional numbness, reduced positivity, weight gain, sexual disfunctions, and suicidal thoughts. Due to the limitations of existing treatments, we believe there is significant unmet need for new therapies to improve quality of life outcomes for patient diagnosed with GAD.
65
Psilocybin as a treatment for generalized anxiety disorder
Psychedelic-assisted psychotherapy may provide rapid, significant, and lasting benefit in treating unipolar depression, depression and anxiety symptoms associated with a terminal illness, and substance misuse. Psilocybin is a psychoactive molecule that occurs naturally in several genera of mushrooms, which primarily acts on the serotonin receptor system, and can modulate states of consciousness, cognition, perception, and mood.
When combined with specialized forms of psychotherapeutic support, psilocybin is well tolerated and can reduce scores on mental health severity assessments. Through the 1950s and 1960s, tens of thousands of individuals participated in psychedelic research. While methodologically limited by modern standards, the findings from many of these studies showed substantial improvements in anxiety, depression and addiction levels, and quality of life.
Following decades of socio-political obstruction to psychedelic treatments, an increasing number of clinical psychedelic trials are now being conducted at highly esteemed institutions around the world, including Imperial College London, John Hopkins University, University of California, and now Monash University, Melbourne, in partnership with us.
Over the past decade, the therapeutic potential of psilocybin in anxiety, depression and addiction has been demonstrated in various academic-sponsored studies. In these studies, psilocybin-assisted psychotherapy, provided a rapid reduction in anxiety and depression symptoms on the day of administration with generally maintained treatment effects at follow-up assessments many months later. These studies have shown psilocybin to be generally well-tolerated, with low toxicity and no serious adverse events reported.
We believe that the following four studies detailed below support psilocybin-assisted therapy for treating anxiety using treatment dosages up to 30mg/70kg:
• New York University, Ross et al 2016 (n=29): Rapid and sustained symptom reduction following psilocybin treatment for anxiety and depression in patients with life-threatening cancer: a randomized controlled trial. Psilocybin produced immediate, substantial, and sustained improvements in anxiety and depression, as well as decreases in cancer-related demoralization and hopelessness, improved spiritual wellbeing, and increased quality of life.
• Imperial College London, Carhart-Harris et al 2018 (n=20): Psilocybin with psychological support for treatment-resistant depression: six-month follow-up. Good tolerability, effect sizes large and symptom improvements appeared rapidly after just two psilocybin treatment sessions and remained significant six months post-treatment in a treatment-resistant cohort.
• University of California, Los Angeles, Grob et al 2011 (n=12): Pilot study of psilocybin treatment for anxiety in patients with advanced-stage cancer. The State-Trait Anxiety Inventory trait anxiety subscale demonstrated a significant reduction in anxiety at one and three months after treatment. There were no clinically significant adverse events with psilocybin.
• John Hopkins University, Griffiths et al 2017 (n=51): Psilocybin produces substantial and sustained decreases in depression and anxiety in patients with life-threatening cancer: a randomized double-blind trial. Large and significant decreases in clinician-rated and self-rated measures of depression, anxiety or mood disturbance, and increase measures of quality of life, life meaning, death acceptance, and optimism.
Two psilocybin research programs for depression have received breakthrough designation from the FDA. A small number of other psilocybin treatment development programs are underway globally. Should the results from any of these research programs be positive, approval of psilocybin-assisted psychotherapy as a prescription treatment could occur within the next five years.
Our investigational psilocybin therapy for Generalized Anxiety Disorder
Our psilocybin therapy combines psilocybin with psychological therapy that has been specifically designed for patients diagnosed with generalized anxiety disorder by a multidisciplinary team of experts lead by Principal Investigator Dr Paul Liknaitzky, along with Co-Investigators Professor Suresh Sundram and Professor Murat Yucel. The wider research team includes experts in psychedelic-assisted therapies, psychometric evaluation, qualitative research, therapist training, and risk management. We are in the process of coordinating two clinical trials as part of our clinical development program, which we hope will lead to a Pre-IND submission in Q3 of 2021, and which is ultimately aimed at FDA approval of our psilocybin therapy administered to patients with GAD.
66
Planned Phase 2 exploratory clinical trial
The protocol for our planned Phase 2 Australian exploratory clinical trial has been completed and we anticipate submitting our research proposal to the human research ethics committee (“HREC”) for approval in Q3 of 2021. HREC approval is required prior to the commencement of patient recruitment in Australia. Dr Paul Liknaitzky has successfully achieved HREC approval for other clinical psilocybin studies in Australia and has successfully acquired regulatory permits and imported psilocybin into Australia.
The study is a Phase 2 randomized triple-blind active-placebo-controlled trial to assess the safety and efficacy of psilocybin-assisted psychotherapy for GAD. It will include 72 participants that will experience two psilocybin or active-placebo dosing sessions and up to 11 non-drug, specialist psychotherapy sessions over a period of 10 weeks. Primary outcomes are safety, efficacy and tolerability, and secondary outcomes are quality of life, functional impairment, and comorbidities. Safety will be assessed by monitoring adverse events including but not limited to liver function tests and scores on the Ultra Brief Checklist of Suicidality. Efficacy will be assessed by comparing the change in Hamilton Anxiety Rating Scale from baseline between the placebo and treatment group. Tolerability will be assessed by comparing the proportion of participants who complete both dosing sessions in the placebo and treatment groups. Secondary endpoints will be assessed by monitoring disability, comorbidity, productivity and quality of life using patient reported outcome measures.
A preliminary analysis of patient data will be conducted by an independent data safety monitoring board after 30 patients have completed primary endpoint assessment. The preliminary analysis will allow the trial investigators to inform the second part of the trial, with an opportunity to adjust certain treatment design parameters to optimize patient outcomes, or terminate the trial based on predefined outcomes and adequate conditional power.
FDA development plan and pre-IND meeting
In February 2021, we formally engaged Camargo Pharmaceuticals LLC, to advise upon and compile the pre-investigational new drug application information package necessary to formally request a pre-IND meeting with FDA. This meeting request will be submitted to the FDA in Q3 2021 and we anticipate that the meeting will occur in Q4 2021. We believe that FDA guidance will provide us with the regulatory clarity and commercial confidence to eventually submit an IND to the FDA and subsequently conduct a Phase 2b pivotal clinical trial partly or wholly in the United States to support our future NDA submission.
Psilocybin therapy protocol
Our psilocybin therapy comprises administration of medication with psychotherapy by mental health professionals that have undergone our specialised therapist training program. Therapy is designed to optimize patient safety and therapeutic outcomes in GAD with specific support before, during and after psilocybin dosing sessions.
Each participant will receive two therapeutic doses of our investigational product, which will be composed of a specified dosage of psilocybin, with psychotherapy before, during and after each dose session. The psychotherapy comprises four distinct phases:
• Preliminary psychotherapy: conducted during the screening stage with key focus on clinical formulation, therapeutic alliance, psychedelic treatment psychoeducation and practical preparation for dosing.
• Preparation psychotherapy: conducted following full enrollment and prior to the first dosing session with a key focus on extending preliminary psychotherapy work, and covering more targeted and GAD-specific psychological and practical preparation for dosing.
• After dosing support: conducted within a week following the preparation session with key focus on trust, suitable mindset, conducive physical setting, and participant-led support. Dosing support is the psychotherapy session.
• Integration psychotherapy: conducted following the dosing sessions, including the day directly following each dosing session, with key focus on sustaining benefits through specific mindful, emotion and somatic-focused therapy, meaning-centered support, and facilitating contextual changes that support outcomes.
Therapist recruitment in anticipation of the Phase 2 exploratory trial has commenced and therapist training is anticipated to commence in Q3 2021.
67
Monash University
In December 2020, we entered into a partnership agreement with Monash University (“Monash”) in Australia to conduct a psilocybin-assisted psychotherapy trial to treat GAD. Monash will sponsor our initial Phase 2 exploratory clinical trial, ensuring rigorous scientific independence and the highest standards in ethical and safe research. We are funding and supporting this investigator-initiated trial, and retain all intellectual property created by the trial. We are also investigating the commencement of other psychedelic medicine research projects that would offer an opportunity to address what we believe is an unmet need in patients diagnosed with other mental illnesses.
Monash is one of Australia’s leading universities and consistently ranks among the world’s top 100. Psychedelic treatment for our exploratory trials will be delivered within BrainPark, a state-of-the-art research platform at Monash’s Turner Institute for Brain and Mental Health and Biomedical Imaging Facility, that provides a highly conducive environment for psychedelic treatments in a research context. Both the School of Psychological Sciences within the Turner Institute for Brain and Mental Health, and the Department of Psychiatry within the School of Clinical Sciences, have combined forces to conduct psychedelic research and the team comprises leading researchers and clinicians in relevant fields of psychiatry, psychotherapy, and mental health treatment development.
Clinical trial investigators
The Principal Investigator is Dr Paul Liknaitzky, with Co-Investigators Professor Murat Yucel and Professor Suresh Sundram.
Dr. Liknaitzky is Head of the Clinical Psychedelic Research Lab within the Turner Institute and the Dept of Psychiatry, Monash. He is a Chief Principal Investigator and Research Fellow at Monash University, and has Adjunct or Honorary appointments at St Vincent’s Hospital, Macquarie University, Deakin University, and the University of Melbourne. He earned an Honours in Neuroscience and a PhD in Psychology from the University of Melbourne. His work examines mechanisms of mental illness and treatment development primarily within mood, anxiety and addiction research. Liknaitzky is an Investigator across a number of Australia’s first clinical psychedelic trials. He has been invited to deliver numerous academic, professional, and public talks on psychedelic-assisted psychotherapy, and has been interviewed on the topic for print media, radio, and podcasts. Liknaitzky leads Australia’s first clinical psychedelic lab, coordinates Australia’s first applied psychedelic therapist training program, and is establishing Australia’s largest psychedelic trial (Psi-GAD). His work is focused on developing a rigorous program of research in psychedelic medicine at Monash University that seeks to evaluate therapeutic effects, innovate on treatment design, mitigate known risks, explore potential drawbacks, and understand therapeutic mechanisms.
Professor Murat Yucel gained a PhD combined with specialist clinical training in Clinical Neuropsychology in 2001 at La Trobe University. He then worked across as numerous mental health research centres at the University of Melbourne and was promoted to professor in 2012. He now works within the Monash School of Psychological Sciences, where he heads the mental health and addiction research programs. He is a director of BrainPark — a world-first neuroscience research clinic designed to bring the latest neuroscience with diagnostic or therapeutic benefit to the community in an accessible way.
Professor Suresh Sundram is the Head, Department of Psychiatry, School of Clinical Sciences, Monash University and Director of Research, Mental Health Program, Monash Health. He has been investigating the molecular pathology of schizophrenia and related psychotic disorders using pharmacological, neurochemical and neuropathological approaches. These inter-related methods have been applied to parse components of the disorder such as treatment resistance and suicide to better understand their neurobiological substrates. He undertook his doctoral and post-doctoral studies at the Mental Health Research Institute in Melbourne before establishing his laboratory there and subsequently at the Florey Institute and concurrently establishing a clinical research laboratory undertaking clinical trial and biomarker research in psychotic disorders. He then transferred to and integrated his research program at Monash University and Monash Medical Centre.
Intellectual Property Strategy
We strategically protect our innovations with a harmonized IP strategy, combining patent protection with regulatory and market exclusivity. We are pursuing patent protection for aspects of our psilocybin therapy program. The patent position that will be available to us is unlikely to cover psilocybin alone as a clinical entity. However, we are pursuing a patent position in relation methods of treatment using psilocybin including combination therapies (e.g., formulations, actives plus psychotherapeutic modalities) and other therapeutic methods (e.g., specific dosage regimens).
68
Intellectual Property
We have implemented a patent filing strategy as we develop our products and therapies in conjunction with our medical advisory board. As of September 20, 2021, we own pending patent applications relating to our cannabinoid drug candidates IHL-42X, IHL-216A and IHL-675A and psilocybin assisted psychotherapy program. A summary of the number of patents, patent types and jurisdictions in listed in the table below. Once converted to the complete/PCT stage, the provisional patents will also be applicable to all PCT contracting states. International search reports and written opinions of the International Search Authority have confirmed that the key claims in our filed Patent Cooperation Treaty applications are novel and inventive and that the invention meets the requirements of Industrial Applicability.
|
Product/technology |
Number of |
Type of patent |
Applicable |
|||
|
IHL-42X/Compositions and methods for the treatment of obstructive sleep apnoea (OSA) |
1 |
Complete/PCT |
All 153 PCT contracting states# |
|||
|
IHL-675A/Compositions and methods for the treatment of an inflammatory conditions |
2 |
Provisional |
Australia |
|||
|
IHL-675A/Compositions and methods for the treatment or prevention of an inflammatory condition |
Complete/PCT |
All 153 PCT contracting states# |
||||
|
IHL-216A/Compositions and methods for the treatment or prevention of traumatic brain injury (TBI) |
1 |
Complete/PCT |
All 153 PCT contracting states# |
|||
|
Psi-GAD/Methods for the treatment of generalized anxiety disorder (GAD) |
1 |
Provisional |
Australia |
____________
# Standard/utility patents derived from the PCT application are intended to be pursued in key jurisdictions including Australia, US, Europe, Japan and Israel.
In addition to pursing patent protection for all of our assets, we rely on unpatented trade secrets, know-how and other confidential information as well as proprietary technological innovation and expertise that are protected in part by confidentiality and invention assignment agreements with our employees, advisors and consultants.
Patent matters in biotechnology are highly uncertain and involve complex legal and factual questions. The availability and breadth of claims allowed in biotechnology and pharmaceutical patents cannot be predicted. Statutory differences in patentable subject matter may limit the scope of protection we can obtain on some or all of our licensed inventions or prevent us from obtaining patent protection either of which could harm our business, financial condition and results of operations. Since patent applications are not published until at least 18 months from their first filing date and the publication of discoveries in the scientific literature often lags behind actual discoveries, we cannot be certain that we, or any of our licensors, were the first creator of inventions covered by pending patent applications, or that we or our licensors, were the first to file patent applications for such inventions. Additionally, the grant and enforceability of a patent is dependent on a number of factors that may vary between jurisdictions. These factors may include the novelty of the invention, the requirement that the invention not be obvious in the light of prior art (including prior use or publication of the invention), the utility of the invention and the extent to which the patent clearly describes the best method of working the invention. In short, this means that claims granted in various territories may vary and thereby influence commercial outcomes.
While we have applied for and will continue to file for protection as appropriate for our therapeutic products and technologies, we cannot be certain that any future patent applications we file, or licensed to us, will be granted, or that we will develop additional proprietary products or processes that are patentable or that we will be able to license any other patentable products or processes. We cannot be certain that others will not independently develop similar products or processes, duplicate any of the products or processes developed or being developed by the company or licensed to us, or design around the patents owned or licensed by us, or that any patents owned or licensed by us will provide us with competitive advantages.
Furthermore, we cannot be certain that patents held by third parties will not prevent the commercialization of products incorporating the technology developed by us or licensed to us, or that third parties will not challenge or seek to narrow, invalidate or circumvent any of the issued, pending or future patents owned or licensed by us.
Our commercial success will also depend, in part, on our ability to avoid infringement of patents issued to others. If a court determines that we were infringing any third-party patents, we could be required to pay damages, alter our products or processes, obtain licenses or cease certain activities. We cannot be certain that the licenses required under patents held by third parties would be made available on terms acceptable to us or at all. To the extent that we are unable to obtain such licenses, we could be foreclosed from the development, export, manufacture or commercialization of
69
the product requiring such license or encounter delays in product introductions while we attempt to design around such patents, and any of these circumstances could have a material adverse effect on our business, financial condition and results of operations. We may have to resort to litigation to enforce any patents issued or licensed to us or to determine the scope and validity of third-party proprietary rights. Such litigation could result in substantial costs and diversion of effort by us. We may have to participate in opposition proceedings before the Australian Patent and Trademark Office or another foreign patent office, or in interference proceedings declared by the United States Patent and Trademark Office, to determine the priority of invention for patent applications filed by competitors. Any such litigation interference or opposition proceeding, regardless of outcome, could be expensive and time consuming, and adverse determinations in any such proceedings could prevent us from developing, manufacturing or commercializing our products and could have a material adverse effect on our business, financial condition and results of operations.
As of June 30, 2021, the Company also owns trademark registrations in Australia, United States, Europe, China and Japan.
As of 20 September, 2021, we added 5 new patent applications to our portfolio: PCT/AU2020/051056, PCT/AU2021/050226, PCT/AU2021/050734, Australian Provisional Patent Application No. AU 2021902170, and Australian Provisional Patent Application No. AU 2021902426.
Patent Portfolio
The following table presents our portfolio of patents and patent applications, including their status (as at 20 September, 2021) and title.
|
Patent Family |
Title |
Status |
Expires |
|||
|
PCT/AU2020/051056 |
Compositions for the treatment or prevention of traumatic brain injury |
Pending |
02/10/2040* |
|||
|
PCT/AU2021/050226 |
Methods and compositions for treating or preventing an inflammatory condition |
Pending |
15/03/2041* |
|||
|
PCT/AU2021/050734 |
Compositions and methods for the treatment of obstructive sleep apnoea (OSA) |
Pending |
09/07/2041* |
|||
|
AU 2021902170 |
A composition and uses thereof |
Pending (Provisional) |
14/07/2042^* |
|||
|
AU 2021902426 |
A method of treatment |
Pending (Provisional) |
04/08/2042^* |
____________
* Expiry date may be subject to any patent term extensions or adjustments that may be available.
^ Estimated expiry date of complete application claiming priority from the pending provisional application
Operations Summary
Unregistered Cannabinoid Products
In February 2019, we launched a line of pharmaceutical grade cannabinoid oil products to treat conditions approved for treatment with cannabinoid under the Special Access Scheme. We sold our cannabinoid oil products under the Special Access Scheme. As of April 1, 2021, we ceased selling cannabinoid oil products to focus on the development of our drug candidates.
Material Contracts
Clinical Trial Research Agreement with Alfred Health, dated June 22, 2021
On June 22, 2021, we entered into a Clinical Trial Research Agreement with Alfred Health. Under the terms of the agreement, Alfred Health is to conduct and manage an open label extension on the examination of the combination of dronabinol and acetazolamide for treatment of OSA. The open label extension is to be conducted on a maximum of 12 study participants. Incannex will provide all the information required to conduct the study and will pay market-standard fees to Alfred Health as consideration for its role. The agreement may be terminated by either party for cause, for the reasons set forth in the agreement, and by Incannex at will upon written notice delivered to Alfred Health thirty days prior to the termination date.
70
Clinical Trial Research Agreement with Alfred Health, dated September 24, 2020
On September 24, 2020, we entered into a Clinical Trial Research Agreement with Alfred Health. Under the terms of the agreement, Alfred Health is to conduct and manage a dose finding crossover trial investigating the effect of dronabinol combined with acetazolamide on AHI in adults with OSA. The dose finding crossover trial is to be conducted on a maximum of 12 trial participants. Incannex will provide all the information required to conduct the study and will pay market-standard fees to Alfred Health as consideration for its role. The agreement may be terminated by either party for cause, for the reasons set forth in the agreement, and by Incannex at will upon written notice delivered to Alfred Health thirty days prior to the termination date.
Clinical Trial Research Agreement with University of Western Australia
On April 6, 2021, we entered into a Clinical Trial Research Agreement with University of Western Australia. Under the terms of the agreement, the University of Western Australia is to conduct and manage a dose finding crossover trial investigating the effect of dronabinol combined with acetazolamide on AHI in adults with OSA. The dose finding crossover trial is to be conducted on a maximum of 12 trial participants. Incannex will provide all the information required to conduct the study and will pay market-standard fees to the University of Western Australia as consideration for its role. The agreement may be terminated by either party for cause, for the reasons set forth in the agreement, and by Incannex at will upon written notice delivered to the University of Western Australia thirty days prior to the termination date.
Master Consultancy Agreement with Clinical Network Services (CNS) Pty Ltd
On June 29, 2020, we entered into a Master Consultancy Agreement with Clinical Network Services (CNS) Pty Ltd (“Clinical Network”). Under the terms of the agreement, Clinical Network is to act as Australian and New Zealand consultant to product development and management of clinical research programs. Incannex will pay market-standard fees to Clinical Network. The agreement may be terminated by either party for cause, for the reasons set forth in the agreement.
Research Services Agreement with Monash University, dated November 27, 2020
On November 27, 2020, we entered into a Research Services Agreement with Monash University. Under the terms of the agreement, Monash University is to conduct research services with respect to Psi-GAD. Research activities are to be conducted with respect to a phase 2A randomized double-blind active-placebo-controlled trial to assess the safety and efficacy of Psi-GAD. Incannex will provide all the information required to conduct the study and will pay market-standard fees to Monash University. The agreement may be terminated by either party for cause, for the reasons set forth in the agreement.
Research Services Agreement between Monash University, dated March 10, 2021
On March 10, 2021, we entered into a Research Services Agreement with Monash University. Under the terms of the agreement, Monash University is to conduct research services with respect to TBI. Research activities are to be conducted with respect to the neuroprotective effect of the combination of CBD and isoflurane in a rodent model of mild traumatic brain injury. Incannex will provide all the information required to conduct the study and will pay market-standard fees to Monash University. The agreement may be terminated by either party for cause, for the reasons set forth in the agreement.
Master Service Agreement between Avance Clinical Pty Limited
On July 12, 2021, we entered into a Master Service Agreement with Avance Clinical Pty Limited (“Avance”). Under the terms of the agreement, Avance will perform services to support Incannex’s clinical trials and studies, as requested by Incannex. the agreement has an initial term of five years. Each party may terminate the agreement by delivering a written notice three months prior to the expiration of the term of the contract.
Appendix No. 2 to the Master Consultancy Agreement with Novotech Australia Pty Limited
On February 2, 2021, we entered into Appendix No. 2 to the Master Consultancy Agreement with Novotech Australia Pty Limited (“Novotech”), an affiliate of Clinical Network. Under the terms of the agreement, Novotech is to conduct an open label extension on the examination of the combination of dronabinol and acetazolamide for treatment of OSA. The terms of this agreement are governed by the terms of the Master Consultancy Agreement entered into with Clinical Network.
71
Quantitative and Qualitative Disclosures about Market Risk
Our cash consist entirely of cash held in interest-bearing accounts with banks in Australia. Thus, our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of Australian interest rates. However, a sudden change in market interest rates would not be expected to have a material impact on our financial condition and/or results of operation.
Competition
We are targeting indications that have no registered, limited or costly pharmacological solutions. Thus, competitor drugs for the indications we are assessing with our drug candidates either do not exist or are limited in efficacy or have unacceptable side effect profiles for certain cohorts of patients. The table below outlines existing drugs and therapies used to treat the illnesses we aim to treat with our drug candidates and their associated pitfalls for patients.
|
IHL Drug Candidate |
Indication |
Existing Products |
Existing Product Pitfalls |
|||
|
IHL-42X |
Obstructive Sleep Apnoea |
− CPAP device |
− Noisy mechanical device worn during sleep; − potential poor patient compliance due to discomfort. |
|||
|
IHL-216A |
Traumatic Brain Injury/Concussion |
None |
N/A |
|||
|
IHL-675A |
Lung Inflammation |
− Corticosteroids − Ventilator |
− Corticosteroids reduce immune system activity; − ventilators are associated with a high rate of mortality. |
|||
|
IHL-675A |
Rheumatoid Arthritis |
− Corticosteroids − DMARDS − Biologic agents |
− High expense, significant side effect profiles; − lack of efficacy or tolerability in certain patient cohorts. |
|||
|
IHL-675A |
Inflammatory Bowel Disease |
− Corticosteroids − Immune system suppressors (ISSs) − Biologic agents |
− Corticosteroids can reduce immune system activity; − ISSs can damage the digestive tract lining; |
|||
|
PSI-GAD |
Generalized Anxiety Disorder |
− Antidepressants (SSRI/SNRI classes) |
− Non-curative, poor side effect profile; − some patients become treatment resistant. |
Regulatory Authorities
The ongoing research and development, clinical, regulatory, commercial and manufacturing activities of our drug candidates are subject to extensive regulation by numerous governmental authorities, including (i) in Australia, principally the Therapeutics Goods Administration, or TGA; (ii) in the United States, principally the Food and Drug Administration, or FDA, as well as the Drug Enforcement Agency (DEA); and (iii) in Europe, principally the European Medicines Agency, or EMA and local competent authorities, ethics committees (ECs), institutional research boards (IRBs) and other regulatory authorities at federal, state or local levels. We, along with our third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval and post-approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our drug candidates.
72
United States
FDA process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. Regulations that govern the pharmaceutical quality, safety, efficacy, labeling, storage, record keeping, advertising and promotion of such products under the Federal Food, Drug and Cosmetic Act, the Public Health Service Act, and their implementing regulations. In particular, controlled substances, like synthetic cannabidiol and THC, are regulated by the U.S. Drug Enforcement Administration, or DEA.
The process of obtaining FDA approval and achieving and maintaining compliance with applicable laws and regulations requires the expenditure of substantial time and financial resources. Failure to comply with applicable FDA or other requirements may result in refusal to approve pending applications, a clinical hold, warning letters, civil or criminal penalties, recall or seizure of products, partial or total suspension of production or withdrawal of the product from the market. FDA approval is required before any new drug or biologic, including a new use of a previously approved drug, can be marketed in the United States. All applications for FDA approval must contain, among other things, information relating to safety and efficacy, pharmaceutical quality, packaging, labeling and quality control.
Government oversight of the pharmaceutical industry is usually classified into pre-approval and post-approval categories. Most of the therapeutically significant innovative products marketed today are the subject of New Drug Applications, or NDAs, or Biologics License Applications, or BLAs. Pre-approval activities are used to assure the product is safe and effective before marketing.
Drug Approval Process — FDA
None of our drug candidates may be marketed in the United States until the drug has received FDA approval. The steps required before a drug may be marketed in the United States generally include the following:
• completion of extensive pre-clinical laboratory tests, animal studies, and formulation studies in accordance with the FDA’s GLP and GMP regulations;
• submission to the FDA of an Investigational New Drug, or IND, application for human clinical testing, which must become effective before human clinical trials may begin;
• receive approval from the DEA prior to commencement of any clinical trials in the United States that involve the use of Schedule I controlled substances.
• performance of adequate and well-controlled human clinical trials in accordance with GCP requirements to establish the safety and efficacy of the drug for each proposed indication;
• submission to the FDA of an NDA/BLA after completion of all pivotal clinical trials;
• satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the active pharmaceutical ingredient, or API, and finished drug product are produced and tested to assess compliance with current Good Manufacturing Practices, or cGMPs; and
• FDA review and approval of the NDA/BLA and DEA scheduling (for a controlled substance) prior to any commercial marketing or sale of the drug in the United States.
The manufacturing and quality, as well as preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot guarantee approval of our drug candidates will be granted on a timely basis, if at all. Notably, the FDA may reach different conclusions than we have after analyzing the same data, or there may difference of opinion amongst members of FDA’s review team.
The FDA may inspect and audit domestic and foreign development facilities, planned production facilities, clinical trial sites and laboratory facilities. There is a pre-approval inspection after submission to market a new product, routine inspection of a regulated facility and a “for-cause” inspection to investigate a specific problem that has come to FDA’s attention. After the product is approved and marketed, the FDA uses different mechanisms for assuring that firms adhere to the terms and conditions of approval described in the application and that the product is manufactured in a consistent and controlled manner. This is done by periodic unannounced inspections of production and quality control facilities by FDA’s field investigators and analysts.
73
Preclinical tests include laboratory evaluation of toxicity in animals and in vitro (laboratory tests). The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND application to the FDA. The IND application is based on the results of initial testing done on animals for pharmacology and toxicity, which is used to develop a plan for testing the drug on humans. Only after preclinical testing, FDA determines whether the drug should be tested in people.
Further, an independent institutional review board, or IRB, covering each medical center proposing to conduct clinical trials must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive Good Clinical Practice, or GCP, regulations, which include requirements that all research subjects provide informed consent and that all clinical studies be conducted under the supervision of one or more qualified investigators.
Clinical trials (under an IND) involve administration of the investigational drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol must be provided to the FDA as part of a separate submission to the IND. Further, an Institutional Review Board, or IRB, for each medical center proposing to conduct the clinical trial must review and approve the study protocol and informed consent information for study subjects for any clinical trial before it commences at that center, and the IRB must monitor the study until it is completed. There are also requirements governing reporting of ongoing clinical trials and clinical trial results to public registries. Study subjects must sign an informed consent form before participating in a clinical trial.
Progress reports detailing the results of the clinical studies must be submitted at least annually to the FDA and safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse reactions in case of an open IND. For purposes of an NDA or BLA submission and approval, human clinical trials are typically conducted in the following sequential phases, which may overlap:
• Phase I: Trials are initially conducted in a limited population of healthy human (in oncology Phase I trials are often conducted in patients) subjects or patients to test the drug candidate for safety and dose tolerance. These studies are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness.
• Phase II: The investigational product is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials.
• Phase III: The investigational product is administered to an expanded patient population in adequate and well-controlled studies to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit relationship of the investigational product and to provide an adequate basis for product approval.
• Phase IV: In some cases, the FDA may condition approval of an NDA or BLA on the sponsor’s agreement to conduct additional clinical trials to further assess the drug candidate’s safety, purity and potency after NDA or BLA approval. Such post-approval trials are typically referred to as Phase IV clinical trials.
Concurrent with clinical studies, sponsors usually complete additional animal studies and must also develop additional information about the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, the manufacturer must develop and validate methods for testing the identity, strength, quality and purity of the final product. Moreover, appropriate packaging must be selected and tested and stability studies must be conducted to assure product integrity and demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
The results of product development, preclinical studies and clinical trials, along with the aforementioned manufacturing information, are submitted to the FDA as part of a BLA/NDA. Under the Prescription Drug User Fee Act, or PDUFA, the FDA agrees to specific goals for NDA/BLA review time. The testing and approval process requires substantial time, effort and financial resources. The FDA will review the BLA/NDA and may deem it to be
74
inadequate to support approval, and we cannot be sure that any approval will be granted on a timely basis, if at all. The FDA may also refer the application to the appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of the advisory committee, but it typically follows such recommendations.
The FDA may deny approval of a BLA/NDA if the applicable regulatory criteria are not satisfied, or it may require additional clinical data or additional pivotal Phase III clinical trials. Even if such data are submitted, the FDA may ultimately decide that the NDA/BLA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than the sponsor does. Once issued, product approval may be withdrawn by the FDA if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase IV clinical trials, and surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs which may include pediatric assessment, and potentially studies required for an application for a new indication, new dosage form, a new dosing regimen, a new route of administration or a new active ingredient. Products may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labeling or manufacturing processes or facilities, approval of a new or supplemental BLA may be required, which may involve conducting additional preclinical studies and clinical trials.
Expedited Review and Approval
The FDA has various programs, including fast track designation, priority review, accelerated approval, and breakthrough therapy designation, which are intended to expedite or simplify the process for reviewing drugs and/or provide for approval on the basis of surrogate endpoints. Even if a drug qualifies for one or more of these programs, the FDA may later decide that the drug no longer meets the conditions for qualification or that the time period for FDA review or approval will not be shortened. In particular, if accelerated approval is granted for any particular clinical product, the FDA can subsequently revoke the marketing authorization for such product if post-market clinical trial results are unsuccessful. Generally, drugs that may be eligible for these programs are those for serious or life-threatening diseases or conditions, those with the potential to address unmet medical needs, and those that offer meaningful benefits over existing treatments.
Other U.S. Regulatory Requirements
After approval, products are subject to extensive continuing regulation by the FDA, which include company obligations to manufacture products in accordance with GMP, maintain and provide to the FDA updated safety and efficacy information, report adverse experiences with the product, keep certain records and submit periodic reports, obtain FDA approval of certain manufacturing or labelling changes and comply with FDA promotion and advertising requirements and restrictions. Failure to meet these obligations can result in various adverse consequences, both voluntary and FDA-imposed, including product recalls, withdrawal of approval, restrictions on marketing, and the imposition of civil fines and criminal penalties against the BLA holder — all of which may become public. In addition, later discovery of previously unknown safety or efficacy issues may result in restrictions on the product, manufacturer or application holder.
We, and any manufacturers of our drug candidates, are required to comply with applicable FDA manufacturing requirements contained in the FDA’s GMP regulations. GMP regulations require, among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation. The manufacturing facilities for our drug candidates must meet GMP requirements. We, and any third-party manufacturers, are also subject to periodic inspections of facilities by the FDA and other authorities, including procedures and operations used in the testing and manufacture of our drug candidates to assess our compliance with applicable regulations.
With respect to post-market product advertising and promotion, the FDA imposes a number of complex regulations on entities that advertise and promote pharmaceuticals, which include, among others, standards for direct-to-consumer advertising, promoting products for uses or in patient populations that are not described in the product’s approved labelling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the Internet. Failure to comply with FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors and civil or criminal penalties. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses.
75
Changes to some of the conditions established in an approved application, including changes in indications, labelling, or manufacturing processes or facilities, require submission and FDA approval of a new BLA or BLA supplement before the change can be implemented. A BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing BLA supplements as it does in reviewing BLAs.
Adverse event reporting and submission of periodic reports is required following FDA approval of a BLA. The FDA also may require post-marketing testing, known as Phase IV testing, risk mitigation strategies, and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product.
Controlled Substances
The CSA and its implementing regulations establish a “closed system” of distribution for controlled substances. The CSA imposes registration, security, recordkeeping and reporting, storage, manufacturing, distribution, labeling, importation, exportation, disposal and other requirements under the oversight of the DEA. The DEA is the federal agency responsible for regulating controlled substances, and requires those individuals or entities that manufacture, import, export, distribute, research, or dispense controlled substances to comply with the regulatory requirements to prevent the diversion of controlled substances to illicit channels of commerce.
Facilities that research, manufacture, distribute, import or export any controlled substance must register annually with the DEA. The DEA registration is specific to the particular location, activity(ies) and controlled substances utilized. For example, separate registrations are required for importation and manufacturing activities, and each registration authorizes which schedules of controlled substances the registrant may handle. However, certain coincident activities are permitted without obtaining a separate DEA registration, such as distribution of controlled substances by the manufacturer that produces them.
The DEA categorizes controlled substances into one of five schedules — Schedule I, II, III, IV, or V — with varying qualifications for listing in each schedule. Scheduling determination by the DEA are dependent on approval of a substance or a specific formulation of a substance. Schedule I substances by definition have a high potential for abuse, have no currently “accepted medical use” in treatment in the United States and lack accepted safety for use under medical supervision. They may be used only in federally-approved research programs and may not be marketed or sold for dispensing to patients in the United States. Pharmaceutical products having a currently accepted medical use may be listed as Schedule II, III, IV or V substances, with Schedule II substances presenting the highest potential for abuse and physical or psychological dependence, and Schedule V substances presenting the lowest relative potential for abuse and dependence. The regulatory requirements are more restrictive for Schedule II substances than Schedule III-V substances. For example, all Schedule II drug prescriptions must be signed by a physician, physically presented to a pharmacist in most situations, and cannot be refilled. Marijuana and THC are Schedule I controlled substances under the CSA. Products approved for medical use in the United States that contain marijuana, THC or marijuana/THC extracts, must be placed in Schedules II-V, since approval by the FDA satisfies the “acceptable medical use” requirement. While marijuana and THC are controlled substances, the Agricultural Improvement Act of 2018 amended the CSA to exclude Cannabis meeting the statutory definition of hemp from the definition of marijuana. As a result, Cannabis that contains 0.3 percent or less of delta-9 THC on a dry weight basis is no longer considered a controlled substance. By extension, Cannabis-derived cannabidiol that satisfies the same limitation concerning delta-9 THC is also excluded from CSA regulatory controls. Because the definition of hemp does not expressly include synthetic equivalents of Cannabis or its derivatives, however, there is a lack of clarity about the CSA control status of pharmaceutically manufactured cannabidiol. Absent guidance to the contrary from the DEA, Cannabis and those products which contain Cannabis, that do not meet the definition of hemp remain in Schedule I of the CSA for purposes of development and research activities.
The DEA inspects all manufacturing facilities to review security, record keeping, reporting and compliance with other DEA regulatory requirements prior to issuing a controlled substance registration. The specific security requirements vary by the type of business activity and the schedule and quantity of controlled substances handled. The most stringent requirements apply to manufacturers of Schedule I and Schedule II substances. Required security measures commonly include background checks on employees and physical control of controlled substances through storage in approved vaults, safes and cages, and through use of alarm systems and surveillance cameras. An application for a manufacturing registration as a bulk manufacturer (not a dosage form manufacturer or a repacker/relabeler) for a Schedule I or II substance must be published in the Federal Register and is open for 30 days to permit
76
interested persons to submit comments, objections, or requests for a hearing. A copy of the notice of the Federal Register publication is forwarded by the DEA to all those registered, or applicants for registration, as bulk manufacturers of that substance. Once registered, manufacturing facilities must maintain records documenting the manufacture, receipt and distribution of all controlled substances. Manufacturers must submit periodic reports to the DEA of the distribution of Schedule I and II controlled substances, Schedule III narcotic substances, and other designated substances. Registrants must also report any controlled substance thefts or significant losses and must adhere to certain requirements to dispose of controlled substances. As with applications for registration as a bulk manufacturer, an application for an importer registration for a Schedule I or II substance must also be published in the Federal Register, which remains open for 30 days for comments. Imports of Schedule I and II controlled substances for commercial purposes are generally restricted to substances not already available from a domestic supplier or where there is not adequate competition among domestic suppliers. In addition to an importer or exporter registration, importers and exporters must obtain a permit for every import or export of a Schedule I and II substance, Schedule III, IV and V narcotic, specially designated Schedule III non-narcotics, or Schedule IV or V narcotic controlled in Schedule I or II by the Convention on Psychotropic Substances and submit import or export declarations for Schedule III, IV and V non-narcotics.
For drugs manufactured in the United States, the DEA establishes annually an aggregate quota for the amount of substances within Schedules I and II that may be manufactured or produced in the United States based on the DEA’s estimate of the quantity needed to meet legitimate medical, scientific, research and industrial needs. This limited aggregate amount of Cannabis that the DEA allows to be produced in the United States each year is allocated among individual companies, which, in turn, must annually apply to the DEA for individual manufacturing and procurement quotas. The quotas apply equally to the manufacturing of the active pharmaceutical ingredient and production of dosage forms. The DEA may adjust aggregate production quotas a few times per year, and individual manufacturing or procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments for individual companies.
The states also maintain separate controlled substance laws and regulations, including licensing, recordkeeping, security, distribution, and dispensing requirements. State Authorities, including Boards of Pharmacy, regulate use of controlled substances in each state. Failure to maintain compliance with applicable requirements, particularly as manifested in the loss or diversion of controlled substances, can result in enforcement action that could have a material adverse effect on our business, operations and financial condition. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to revoke those registrations. In certain circumstances, violations could lead to criminal prosecution.
We will not be subject to the DEA approval to conduct our clinical trials for the foreseeable future because we have conducted and plan to continue to conduct clinical trials for each clinical drug program in Australia. We may also decide to develop, manufacture or commercialize our drug candidates in additional countries. As a result, we will be subject to controlled substance laws and regulations from the TGA in Australia, Health Canada’s Office of Controlled Substances in Canada, the Drugs & Firearms Unit (Home Office) of the National Drug Control System in the United Kingdom, and from other regulatory agencies in other countries where we develop, manufacture or commercialize each drug asset in the future.
Foreign Regulation
In addition to regulations in the United States, we are subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our drug candidates. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials vary greatly from country to country.
European Union and United Kingdom
In the European Economic Area, or EEA, which is comprised of the Member States of the European Union plus Norway, Iceland and Liechtenstein, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA by the EMA. Under European Union regulatory systems, we must submit and obtain authorization for a clinical trial application in each member state in which we intend to conduct a clinical trial. When conducting clinical trials in the European Union, we must adhere to the provisions of the European Union Clinical Trials Directive (Directive 2001/20/EC) and the laws and regulations of the EU Member States implementing them.
77
These provisions require, among other things, that the prior authorization of an Ethics Committee and the competent Member State authority is obtained before commencing the clinical trial. In April 2014, the European Union passed the Clinical Trials Regulation (Regulation 536/2014), which will replace the current Clinical Trials Directive. To ensure that the rules for clinical trials are identical throughout the European Union, the EU Clinical Trials Regulation was passed as a regulation that is directly applicable in all EU member states. All clinical trials performed in the European Union are required to be conducted in accordance with the Clinical Trials Directive until the Clinical Trials Regulation becomes applicable. According to the current plans of the EMA, the Clinical Trials Regulation is expected to become applicable.
After we have completed our clinical trials, we must obtain marketing authorization before we can market our product. We may submit applications for marketing authorizations under a centralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states.
The European Medicines Agency, or EMA, is a body of the European Union located in Amsterdam. The EMA is responsible for the scientific evaluation of medicines developed by pharmaceutical companies for use in the European Union. The EMA is involved in the scientific evaluation of medicines that fall within the scope of the centralized procedure. Like the FDA there is a harmonization between regulators and the EMA may inspect and audit the development facilities, planned production facilities, clinical trial sites and laboratory facilities. Additionally, after the product is approved and marketed, the EMA uses different mechanisms for assuring that firms adhere to the terms and conditions of approval described in the application and that the product is manufactured in a consistent and controlled manner. This is done by periodic unannounced inspections of production and quality control facilities.
If any of our drug candidates receive marketing approval in the EEA, we expect they will benefit from 8 years of data protection and 10 years of market protection. The periods run in parallel so effectively 8 years of data protection plus 2 years of market protection is granted. This means that a biosimilar application referencing our safety and efficacy data held on file at the EMA cannot be filed until the end of the data protection period of 8 years, and the biosimilar cannot be placed on the market until after a further 2 years have elapsed (8 + 2). Furthermore, an additional 1 year of market protection is available (8 + 2 + 1) where we obtain approval of a second indication having a significant clinical benefit in the initial 8-year period.
Similarly, since the Biologics Price Competition and Innovation Act (BPCIA) came into force in 2010, the United States provides 4 years of data exclusivity and 12 years of marketing exclusivity for a new biologic. The periods of exclusivity run in parallel, meaning that the FDA will not accept a biosimilar filing for 4 years and will not approve the biosimilar for a further 8 years (4 + 8).
Regulation and Procedures Governing Approval of Medicinal Products in the European Union
In order to market any product outside of the United States, a company must also comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of products. Whether or not it obtains FDA approval for a product, an applicant will need to obtain the necessary approvals by the comparable foreign regulatory authorities before it can initiate clinical trials or marketing of the product in those countries or jurisdictions. Specifically, the process governing approval of medicinal products in the European Union generally follows the same lines as in the United States, although the approval of a medicinal product in the United States is no guarantee of approval of the same product in the European Union, either at all or within the same timescale as approval may be granted in the United States. It entails satisfactory completion of pharmaceutical development, non-clinical studies and adequate and well-controlled clinical trials to establish the safety and efficacy of the medicinal product for each proposed indication. It also requires the submission to relevant competent authorities for clinical trials authorization for a marketing authorization application, or MAA, and granting of a marketing authorization by these authorities before the product can be marketed and sold in the European Union or its member states (as well as Iceland, Norway and Liechtenstein). If we fail to comply with applicable requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Clinical Trial Approval
Pursuant to the currently applicable Clinical Trials Directive 2001/20/EC and the Directive 2005/28/EC on GCP, a system for the approval of clinical trials in the European Union has been implemented through national legislation of the member states. Under this system, an applicant must obtain approval from the national competent
78
authority, or NCA, of a European Union member state in which the clinical trial is to be conducted or in multiple member states if the clinical trial is to be conducted in a number of member states. Furthermore, the applicant may only start a clinical trial at a specific study site after the independent ethics committee, or EC, has issued a favorable opinion in relation to the clinical trial. The clinical trial application must be accompanied by an investigational medicinal product dossier with supporting information prescribed by Directive 2001/20/EC and Directive 2005/28/EC and corresponding national laws of the member states and further detailed in applicable guidance documents. Under the current regime all suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial have to be reported to the NCA and ECs of the member state where they occurred.
In April 2014, the European Union adopted a new Clinical Trials Regulation (EU) No 536/2014, which is set to replace the current Clinical Trials Directive 2001/20/EC. It will overhaul the current system of approvals for clinical trials in the European Union. Specifically, the new legislation, which will be directly applicable in all EU member states (meaning that no national implementing legislation in each European Union member state is required), aims at simplifying and streamlining the approval of clinical trials in the European Union. For instance, the new Clinical Trials Regulation provides for a streamlined application procedure via a single-entry point and strictly defined deadlines for the assessment of clinical trial applications. It is expected that the new Clinical Trials Regulation will come into effect following confirmation of full functionality of the Clinical Trials Information System, the centralized EU portal and database for clinical trials foreseen by the new Clinical Trials Regulation, through an independent audit, which is currently expected to occur in December 2021.
Marketing Authorization
To obtain a marketing authorization for a product in the European Economic Area (comprised of the EU member states plus Norway, Iceland and Liechtenstein), or EEA, an applicant must submit a MAA, either under a centralized procedure administered by the EMA or one of the procedures administered by competent authorities in the EEA member states (decentralized procedure, national procedure, or mutual recognition procedure). A marketing authorization may be granted only to an applicant established in the EEA.
The centralized procedure provides for the grant of a single marketing authorization by the European Commission that is valid throughout the EEA. Pursuant to Regulation (EC) No 726/2004, our investigational COMP360 psilocybin therapy, as a new active substance indicated for the treatment of treatment-resistant depression, will have the option to be filed through the centralized procedure. For those products for which the use of the centralized procedure is not mandatory, applicants may elect to use the centralized procedure where either the product contains a new active substance indicated for the treatment of diseases other than those on the mandatory list, or where the applicant can show that the product constitutes a significant therapeutic, scientific or technical innovation, or for which a centralized process is in the interest of public health.
Under the centralized procedure, the Committee for Medicinal Products for Human use, or the CHMP, which is the EMA’s committee that is responsible for human medicines, established at the EMA is responsible for conducting the assessment of whether a medicine meets the required quality, safety and efficacy requirements, and whether it has a positive risk/benefit/risk profile. Under the centralized procedure, the maximum timeframe for the evaluation of a MAA is 210 days from the receipt of a valid MAA, excluding clock stops when additional information or written or oral explanation is to be provided by the applicant in response to questions of the CHMP. Clock stops may extend the timeframe of evaluation of a MAA considerably beyond 210 days. Where the CHMP gives a positive opinion, it provides the opinion together with supporting documentation to the European Commission, who make the final decision to grant a marketing authorization. Accelerated evaluation may be granted by the CHMP in exceptional cases, when a medicinal product is of major interest from the point of view of public health and, in particular, from the viewpoint of therapeutic innovation. If the CHMP accepts such a request, the timeframe of 210 days for assessment will be reduced to 150 days (excluding clock stops), but it is possible that the CHMP may revert to the standard time limit for the centralized procedure if it determines that the application is no longer appropriate to conduct an accelerated assessment.
PRIME Scheme
EMA now offers a scheme that is intended to reinforce early dialogue with, and regulatory support from, EMA in order to stimulate innovation, optimize development and enable accelerated assessment of PRIority MEdicines, or PRIME. It is intended to build upon the scientific advice scheme and accelerated assessment procedure offered by EMA. The scheme is voluntary and eligibility criteria must be met for a medicine to qualify for PRIME.
79
The PRIME scheme is open to medicines under development and for which the applicant intends to apply for an initial marketing authorization application through the centralized procedure. Eligible products must target conditions for which where is an unmet medical need (there is no satisfactory method of diagnosis, prevention or treatment in the European Union or, if there is, the new medicine will bring a major therapeutic advantage) and they must demonstrate the potential to address the unmet medical need by introducing new methods or therapy or improving existing ones. Applicants will typically be at the exploratory clinical trial phase of development and will have preliminary clinical evidence in patients to demonstrate the promising activity of the medicine and its potential to address to a significant extent an unmet medical need. In exceptional cases, applicants from the academic sector or SMEs (small and medium sized enterprises) may submit an eligibility request at an earlier stage of development if compelling non-clinical data in a relevant model provide early evidence of promising activity, and first in man studies indicate adequate exposure for the desired pharmacotherapeutic effects and tolerability.
If a medicine is selected for the PRIME scheme, EMA:
• appoints a rapporteur from the CHMP or from the Committee for Advanced Therapies (CAT) to provide continuous support and to build up knowledge of the medicine in advance of the filing of a marketing authorization application;
• issues guidance on the applicant’s overall development plan and regulatory strategy;
• organizes a kick-off meeting with the rapporteur and experts from relevant EMA committees and working groups;
• provides a dedicated EMA contact person; and
• provides scientific advice at key development milestones, involving additional stakeholders, such as health technology assessment bodies and patients, as needed.
Medicines that are selected for the PRIME scheme are also expected to benefit from EMA’s accelerated assessment procedure at the time of application for marketing authorization. Where, during the course of development, a medicine no longer meets the eligibility criteria, support under the PRIME scheme may be withdrawn.
Pediatric Development
In the EEA, companies developing a new medicinal product must agree upon a Pediatric Investigation Plan, or PIP, with the EMA’s Pediatric Committee, or PDCO, and must conduct pediatric clinical trials in accordance with that PIP, unless a waiver applies, (e.g., because the relevant disease or condition occurs only in adults). The PIP sets out the timing and measures proposed to generate data to support a pediatric indication of the drug for which marketing authorization is being sought. The marketing authorization application for the product must include the results of pediatric clinical trials conducted in accordance with the PIP, unless a waiver applies, or a deferral has been granted by the PDCO of the obligation to implement some or all of the measures of the PIP until there are sufficient data to demonstrate the efficacy and safety of the product in adults, in which case the pediatric clinical trials must be completed at a later date. Products that are granted a marketing authorization with the results of the pediatric clinical trials conducted in accordance with the PIP are eligible for a six-month extension of the protection under a supplementary protection certificate (if any is in effect at the time of approval) even where the trial results are negative. In the case of orphan medicinal products, a two-year extension of the orphan market exclusivity may be available. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the PIP are developed and submitted.
Regulatory Data Protection in the European Union
In the EEA, innovative medicinal products approved on the basis of a complete independent data package qualify for eight years of data exclusivity upon grant of a marketing authorization and an additional two years of market exclusivity pursuant to Regulation (EC) No. 726/2004, as amended, and Directive 2001/83/EC, as amended. Data exclusivity prevents generic and biosimilar applicants from referencing the innovator’s preclinical and clinical trial data contained in the dossier of the reference product when applying for a marketing authorization for a period of eight years from the date on which the reference product was first authorized in the EEA. During the additional two-year period of market exclusivity, a generic or biosimilar marketing authorization application can be submitted, and the innovator’s data may be referenced, but no generic or biosimilar medicinal product can be marketed until the
80
expiration of the market exclusivity period. The overall 10-year period will be extended to a maximum of 11 years if, during the first eight years of those 10 years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to authorization, is held to bring a significant clinical benefit in comparison with existing therapies. Even if a compound is considered to be an innovative medicinal product so that the innovator gains the prescribed period of data exclusivity, another company may market another version of the product if such company obtained marketing authorization based on a MAA with a completely independent data package of pharmaceutical tests, preclinical tests and clinical trials.
Periods of Authorization and Renewals
A marketing authorization is valid for five years, in principle, and it may be renewed after five years on the basis of a re-evaluation of the risk benefit balance by the EMA or by the competent authority of the authorizing member state. To that end, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least nine months before the marketing authorization ceases to be valid. Once renewed, the marketing authorization is valid for an unlimited period, unless the European Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal period. Any authorization that is not followed by the placement of the product on the EEA market (in the case of the centralized procedure) or on the market of the authorizing member state within three years after authorization ceases to be valid.
Controlled Drugs Classification
The position in the member states of the European Union is not harmonized. Member states have implemented the relevant UN Conventions (the Single Convention of Narcotic Drugs 1961 and the Convention on Psychotropic Substances 1971) into their national legislation, which has led to differences in how controlled substances are regulated in different countries of the European Union. It is therefore important to determine at a national level whether a substance is controlled and to comply with the applicable legal requirements.
Regulatory Requirements After Marketing Authorization
Following approval, the holder of the marketing authorization is required to comply with a range of requirements applicable to the manufacturing, marketing, promotion and sale of the medicinal product.
These include compliance with the European Union’s stringent pharmacovigilance or safety reporting rules, pursuant to which post-authorization studies and additional monitoring obligations can be imposed. The holder of a marketing authorization must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance, who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports, or PSURs.
In addition, all new MAAs must include a risk management plan, or RMP, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the marketing authorization. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies. RMPs and PSURs are routinely available to third parties requesting access, subject to limited redactions.
Furthermore, the manufacturing of authorized products, for which a separate manufacturer’s license is mandatory, must also be conducted in strict compliance with the EMA’s cGMP requirements and comparable requirements of other regulatory bodies in the European Union, which mandate the methods, facilities and controls used in manufacturing, processing and packing of products to assure their safety and identity.
Finally, the marketing and promotion of authorized products, including industry-sponsored continuing medical education and advertising directed toward the prescribers of products, are strictly regulated in the European Union under Directive 2001/83/EC, as amended. The advertising of prescription-only medicines to the general public is not permitted in the European Union, or in the UK under the Human Medicines Regulations 2021. Although general requirements for advertising and promotion of medicinal products are established under EU Directive 2001/83/EC as amended, the details are governed by regulations in each European Union member state (as well as Iceland, Norway and Liechtenstein) and can differ from one country to another.
81
United Kingdom
The United Kingdom (UK) has left the European Union and will declare its independent processes to approve clinical research and marketing authorizations. Currently, the UK is in a transition period after it left the European Union, leaving EU regulations and agreements active. This transition period ended on December 31, 2020. Since the regulatory framework in the UK covering quality, safety and efficacy of pharmaceutical products, clinical trials, marketing authorization, commercial sales and distribution of pharmaceutical products is derived from EU Directives and Regulations, Brexit could materially impact the future regulatory regime which applies to products and the approval of drug candidates in the UK, as UK legislation now has the potential to diverge from EU legislation. It remains to be seen how Brexit will impact regulatory requirements for drug candidates and products in the UK in the long-term. The MHRA has recently published detailed guidance for industry and organizations to follow from January 1, 2021, which will be updated as the UK’s regulatory position on medicinal products evolves over time. How precisely clinical research within the UK will be performed and how approval for drugs will be organized is subject to ongoing discussions
The UK will no longer be covered by centralized marketing authorizations (under the Northern Irish Protocol, centralized marketing authorizations will continue to be recognized in Northern Ireland). All medicinal products with a current centralized marketing authorization were automatically converted to Great Britain marketing authorizations on January 1, 2021. For a period of two years from January 1, 2021, the MHRA may rely on a decision taken by the European Commission on the approval of a new marketing authorization in the centralized procedure, in order to more quickly grant a new Great Britain marketing authorization. A separate application will, however, still be required.
Australia
In Australia, the relevant regulatory body responsible for the pharmaceutical industry is the Therapeutics Goods Administration, or TGA. As with the EMA and FDA there is a harmonization and collaboration between regulatory authorities. The TGA requires notification of all clinical trials via an electronic submission of a Clinical Trial Notification (CTN) prior to commencing the clinical trial.
Third-Party Payer Coverage and Reimbursement
Although our drug candidates have not been commercialized for any indication, if they are approved for marketing, commercial success of our drug candidates will depend, in part, upon the availability of coverage and reimbursement from third party payers at the federal, state and private levels.
In the United States and internationally, sales of any product that we market in the future, and our ability to generate revenues on such sales, are dependent, in significant part, on the availability of adequate coverage and reimbursement from third party payors, such as state and federal governments, managed care providers and private insurance plans.
Private insurers, such as health maintenance organizations and managed care providers, have implemented cost-cutting and reimbursement initiatives and likely will continue to do so in the future. These include establishing formularies that govern the drugs and biologics that will be offered and the out-of-pocket obligations of member patients for such products. We may need to conduct pharmacoeconomic studies to demonstrate the cost-effectiveness of our drug candidates for formulary coverage and reimbursement. Even with such studies, our drug candidates may be considered less safe, less effective or less cost-effective than existing products, and third-party payors may not provide coverage and reimbursement for our drug candidates, in whole or in part. In addition, particularly in the United States and increasingly in other countries, we are required to provide discounts and pay rebates to state and federal governments and agencies in connection with purchases of our drug candidates that are reimbursed by such entities. It is possible that future legislation in the United States and other jurisdictions could be enacted to potentially impact reimbursement rates for the drug candidates we are developing and may develop in the future and could further impact the levels of discounts and rebates paid to federal and state government entities. Any legislation that impacts these areas could impact, in a significant way, our ability to generate revenues from sales of drug candidates that, if successfully developed, we bring to market. Political, economic and regulatory influences are subjecting the healthcare industry in the United States to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our future business.
82
Similar political, economic and regulatory developments are occurring in the European Union and may affect the ability of pharmaceutical companies to profitably commercialize their products. In addition to continuing pressure on prices and cost containment measures, legislative developments at the European Union or member state level may result in significant additional requirements or obstacles. The delivery of healthcare in the European Union, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than European Union, law and policy. National governments and health service providers have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most EU member states have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing EU and national regulatory burdens on those wishing to develop and market products, this could restrict or regulate post-approval activities and affect the ability of pharmaceutical companies to commercialize their products. In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. In the future, there may continue to be additional proposals relating to the reform of the healthcare system in the United States and international healthcare systems. Future legislation, or regulatory actions implementing recent or future legislation may have a significant effect on our business. Our ability to successfully commercialize products depends in part on the extent to which reimbursement for the costs of our drug candidates and related treatments will be available in the United States and worldwide from government health administration authorities, private health insurers and other organizations. The adoption of certain proposals could limit the prices we are able to charge for our drug candidates, the amounts of reimbursement available for our drug candidates, and limit the acceptance and availability of our drug candidates. Therefore, substantial uncertainty exists as to the reimbursement status of newly approved health care products by third party payors.
Exchange Controls
Australia has largely abolished exchange controls on investment transactions. The Australian dollar is freely convertible into U.S. dollars. In addition, there are currently no specific rules or limitations regarding the export from Australia of profits, dividends, capital or similar funds belonging to foreign investors, except that certain payments to non-residents must be reported to the Australian Cash Transaction Reports Agency, which monitors such transaction, and amounts on account of potential Australian tax liabilities may be required to be withheld unless a relevant taxation treaty can be shown to apply.
The Foreign Acquisitions and Takeovers Act 1975
Under Australian law, in certain circumstances foreign persons are prohibited from acquiring more than a limited percentage of the shares in an Australian company without approval from the Australian Treasurer. These limitations are set forth in the Australian Foreign Acquisitions and Takeovers Act, or the Takeovers Act.
Under the Foreign Acquisitions and Takeovers Act, as currently in effect, any foreign person, together with associates, or parties acting in concert, is prohibited from acquiring 20% or more of the shares in any company having consolidated total assets of or that is valued at A$275 million or more (or A$1,192 million or more in case of U.S. investors). “Associates” is a broadly defined term under the Takeovers Act 1975 and includes the following, but not limited to:
• any relative of the person;
• any person with whom the person is acting or proposes to act in concert;
• any person with whom the person carries on a business in partnership;
• any entity of which the person is a ‘senior officer’ (such as a director or executive);
• if the person is an entity, any holding entity or any senior officer of the entity;
• any entity whose senior officers are accustomed or obliged to act in accordance with the directions, instructions or wishes of the person or if the person is an entity, its senior officers or vice versa;
• any corporation in which the person holds a ‘substantial interest’ (i.e., 20%) or any person holding a substantial interest in the person if a corporation;
83
• a trustee of a trust in which the person holds a substantial interest or if the person is the trustee of a trust, a person who holds a substantial interest in the trust; and
• if the person is a foreign government, a separate government entity or a foreign government investor in relation to a foreign country, any other person that is a foreign government, a separate government entity or foreign government investor, in relation to that country.
The Australian Treasurer also has power in certain circumstances to make an order specifying that two or more persons are associates.
In addition, a foreign person may not acquire shares in a company having consolidated total asset of or that is valued at A$275 million or more (or A$1,192 million or more in case of U.S. investors) if, as a result of that acquisition, the total holdings of all foreign persons and their associates will exceed 40% in aggregate without the approval of the Australian Treasurer. If the necessary approvals are not obtained, the Treasurer may make an order requiring the acquirer to dispose of the shares it has acquired within a specified period of time. The same rule applies if the total holdings of all foreign persons and their associates already exceeds 40% and a foreign person (or its associate) acquires any further shares, including in the course of trading in the secondary market of the ADSs. At present, we do not have total assets of A$275 million or more. At this time, our total assets do not exceed any of the above thresholds and therefore no approval would be required from the Australian Treasurer. Nonetheless, if our total assets were to exceed the threshold in the future, we would be mindful of the number of ADS that can be made available, and monitor the 40% aggregate shareholding threshold for foreign persons (together with the associates) to ensure that it will not be exceeded subject to the Australian Treasurer’s approval.
Each foreign person seeking to acquire holdings in excess of the above caps (including their associates, as the case may be) would need to complete an application form setting out the proposal and relevant particulars of the acquisition/shareholding. The Australian Treasurer then has 30 days to consider the application and make a decision. However, the Australian Treasurer may extend the period by up to a further 90 days by publishing an interim order. The Australian Treasurer has issued a guideline titled Australia’s Foreign Investment Policy which provides an outline of the policy. As for the risk associated with seeking approval, the policy provides that the Treasurer will reject an application if it is contrary to the national interest.
If the level of foreign ownership exceeds 40% at any time, we would be considered a foreign person under the Foreign Acquisitions and Takeovers Act. In such event, we would be required to obtain the approval of the Australian Treasurer for our company, together with our associates, to acquire (i) more than 15% of an Australian company or business having total assets of or that is valued at A$275 million (or A$1,192 if the investor is a non-government entity from a ‘partner agreement’ country) or more; or (ii) any direct or indirect ownership in Australian residential real estate and certain non-residential real estate.
The percentage of foreign ownership in our company may also be included determining the foreign ownership of any Australian company or business in which we may choose to invest. Since we have no current plans for any such acquisition and do not own any property, any such approvals required to be obtained by us as a foreign person under the Takeovers Act will not affect our current or future ownership or lease of property in Australia.
Our Constitution does not contain any additional limitations on a non-resident’s right to hold or vote our securities.
Australian law requires the transfer of shares in our company to be made in writing or electronically through the Clearing House Electronic Sub-register System.
Inflation and Seasonality
Management believes inflation has not had a material impact on our operations or financial condition. Management further believes that our operations are not currently subject to seasonal influences due to our current lack of marketed products. Moreover, the targets of our drug candidates, are not seasonal diseases. Accordingly, once we have marketed products, management does not expect that our business will be subject to seasonal influences.
84
Manufacturing and Raw Materials
We have no manufacturing capabilities and are dependent on third parties for cost effective manufacture and manufacturing process development of our drug candidates. Problems with third party manufacturers or the manufacturing process as such may delay or jeopardize clinical trials and commercialization of our drug candidates.
History and Development
Our legal name is Incannex Healthcare Limited. We were incorporated in Australian in April 2001 under the name Mount Magnet South Limited. In November 2016, we changed name to Impression Healthcare Limited, and in June 2020, to Incannex Healthcare Limited. Incannex is listed on the ASX under the symbol “IHL.”
Since 2019, we have been conducting research and development for medicinal synthetic cannabidiol pharmaceutical products and psychedelic medicine therapies for treatment of a range of indications.
In June 2020, we discontinued the sale of mouthguards for sports activities to focus its resources on cannabinoid sales and development activities. As a result, on June 30, 2020, we sold our wholly-owned subsidiary Gameday International Pty Ltd. Sales of sports mouthguards had severely diminished due to the cancellation of sport seasons resulting from COVID- 19 restrictions. We did not expect any normal continuation of the sports season or recovery in mouthguard sales in the medium term that justifies continued financial commitment.
In January 2019, the Department of Health of Victoria granted us licenses to sell or supply cannabinoid substances, and in particular cannabis, cannabidiol (“CBD”), tetrahydrocannabinols (“THC”) and dronabinol.
Our registered office is located at Suite 15, Level 12, 401 Docklands Drive, Docklands 3008, Victoria, Australia and our telephone number is +61 409 840 786. Our agent for service of process in the United States is Vcorp Agent Services, Inc., 25 Robert Pitt Drive, Suite 204, Monsey, NY 10952, Rockland County. Our address on the Internet is www. incannex.com.au. The information on, or accessible through, our website is not part of this Registration Statement. We have included our website address in this Registration Statement solely as an inactive textual reference. All information we file with the U.S. Securities and Exchange Commission (“SEC”) is available through the SEC’s Electronic Data Gathering
Organizational Structure
Below is a list of our significant subsidiaries, including our ownership percentage, its date of formation and its jurisdiction. These subsidiaries were established to allow us to conduct commercial and clinical operations in Europe and the United States and expand our operations in Australia.
|
Subsidiary |
Ownership |
Date of |
Jurisdiction |
|||
|
Incannex Pty Ltd |
100% |
November 30, 2018 |
Victoria, Australia |
|||
|
Psychennex Pty Ltd |
100% |
November 20, 2020 |
Victoria, Australia |
Property, Plants and Equipment
We own computer equipment, office furniture and laboratory equipment, which is primarily placed at our own offices and laboratories.
|
Office Location |
Lease expiry |
|
|
Suite 15, Level 12, 401 Docklands Drive, Docklands 3008, Victoria, Australia |
April 2022 |
|
|
Suite 207, 11 Solent Circuit, Norwest 2153, NSW, Australia |
September 2021 |
85
Directors and Senior Management
The following table sets forth our directors and senior management, their age and the positions they held as of June 30, 2021. There are no family relationships among any of the members of our board of directors and our senior management.
|
Name |
Age |
Position |
||
|
Joel Latham |
32 |
Chief Executive Officer and Managing Director |
||
|
Troy Valentine(1) |
48 |
Chairman |
||
|
Dr. Sud Agarwal |
46 |
Chief Medical Officer and Director |
||
|
Peter Widdows(2) |
55 |
Director |
||
|
Madhukar Bhalla |
63 |
Chief Financial Officer and Company Secretary |
____________
(1) Member of the Audit Committee.
(2) Chair of Audit Committee.
Joel Latham. Joel Latham has been the Chief Executive Officer and Managing Director of Incannex since July 2018. Mr. Latham is responsible for the Company’s commercial operations, strategic decision-making, and oversight of all clinical development assets for Incannex Healthcare. Prior to his appointment as Chief Executive Officer, Mr. Latham had been a key member of our senior leadership team acting as General Manager since 2016. During this time, he was instrumental in the marketing and procurement of multiple revenue-generating opportunities and partnerships, including with Pacific Smiles (ASX:PSQ), 1300 Smiles (ASX: ONT), the National Rugby League, the Australian Football League, ONE Fighting Championship, FIT Technologies and Cannvalate. During his time at the Company, Mr. Latham has been pivotal in the development and execution of Incannex’s drug development and regulatory strategy. Prior to joing Incannex in 2016, Mr. Latham had over 14 years’ experience, with major firms such as Mars Foods, Tabcorp and Philip Morris International in management and commercial operational roles.
Troy Valentine. Troy Valentine has been Chairman of the Board of Directors since December 2017. Mr. Valentine is a finance professional with managerial and Board experience spanning over 27 years. He commenced his career with Australian brokerage firm Hartley Poynton (now Euroz Hartley’s Limited) in 1994 before moving to Patersons Securities (now Canaccord Genuity) in 2000 and subsequently became an Associate Director. During his time at Patersons, he was responsible for managing both retail and institutional accounts. Mr. Valentine has significant corporate and capital raising experience, especially with start-ups and small to mid-cap size companies. He is currently a director of Australian boutique corporate advisory firm Alignment Capital Pty Ltd, which he co-founded in 2014
Dr. Agarwal has been our Chief Medical Officer of Incannex since June 2019. He is responsible for the oversight over the Company’s cannabinoid clinical program and pipeline of proprietary products. Dr Agarwal is a specialist anaesthesiologist and physician researcher and passed his board exams and was made a Fellow of the Australian and New Zealand College of Anaesthetists in 2009. Dr. Sud Agarwal is a key opinion leader in the clinical use of medicinal cannabis and is regularly invited as a keynote to industry and pharmaceutical events, including the World Cannabis Conference (June 2019), the Australian Medicinal Cannabis Conference (March 2019), Prohibition Partners (September 2020) and the forthcoming International Cannabinoid Derived Pharmaceuticals Summit in Boston (September 2021). Since 2018, Dr. Agarwal also serves as Chief Executive Officer and Chairman of Cannvalate, an Australian private medicinal cannabis company that owns a 3% beneficial interest in Incannex.
Peter Widdows. Peter Widdows has been a Director since 2018. He is a Fellow Chartered Accountant with experience across various functions of business. He has extensive experience in Australian and international consumer goods markets and has worked as a senior executive in numerous geographies, including Europe, the United States and Asia Pacific. In particular, Mr. Widdows served as the Regional Chief Executive Officer — Australasia and Greater China at the H. J. Heinz Company from 2008 to 2010 and as the Chief Executive Officer and Managing Director — Australia at the H. J. Heinz Company from 2002 to 2008 and as the General Manager Strategy & Planning at Starkist Foods Inc. in Cincinnati from 1998 to 2000. Since September 2018, Mr. Widdows has been Chairman of Sunny Queen Australia Ltd, Australia’s largest shell egg and egg-based meal producer and is also a Non-Executive Director of Youi Insurance Holdings Ltd, an Australian general insurance company.
86
Madhukar Bhalla. Madhukar Bhalla has been Chief Financial Officer and Company Secretary of Incannex since June 2021. Since July 2018, he has been acting as Company Secretary and Corporate Administrator at Classic Minerals Limited, an ASX-listed Australian company. Since July 2017, Mr Bhalla has been acting as Company Secretary of Appsolute Digital Ltd, a public unlisted Australian company. Between November 2017 and July 2018, Mr. Bhalla acted as Corporate Governance and HR Manager at Role Models and Leaders Australia and, from 2016 to 2018, he acted as the Company Secretary for FairStar Resources Limited.
Compensation
Remuneration Principles
Remuneration of all executive and non-executive directors and officers is determined by the board of directors.
We are committed to remunerating senior executives and executive directors in a manner that is market-competitive and consistent with “Best Practice” including the interests of shareholders. Remuneration packages are based on fixed and variable components, determined by the executives’ position, experience and performance, and may be satisfied via cash or equity.
Non-executive directors are remunerated out of the aggregate amount approved by shareholders and at a level that is consistent with industry standards. Non-executive directors do not receive performance-based bonuses and prior shareholder approval is required to participate in any issue of equity. No retirement benefits are payable other than statutory superannuation, if applicable.
Our remuneration policy is not directly based on our financial performance, rather on industry practice, given we operate in the biotechnology sector and our primary focus is research activities with a long-term objective of developing and commercializing the research and development results.
We envisage our performance in terms of earnings will remain negative while we continue in the research and development phase.
The purpose of a performance bonus is to reward individual performance in line with our objectives. Consequently, performance-based remuneration is paid to an individual where the individual’s performance clearly contributes to a successful outcome. This is regularly measured in respect of performance against key performance indicators.
We use a variety of key performance indicators to determine achievement, depending on the role of the executive being assessed. These include:
• successful contract negotiations;
• achievement of research project milestones within scheduled time and/or budget; and
• our share price reaching a targeted level on the ASX over a period of time.
87
Executive Compensation
The following table sets forth all of the compensation awarded to, earned by or paid to each individual who served as directors and executive officers in fiscal 2021.
|
June 30, 2021 |
Short-term Benefits |
Post Employment Benefits |
Long-term (share based payments) |
Total |
||||||||
|
Cash salary |
Cash |
Non |
Super- annuation A$ |
Performance Rights, Shares |
A$ |
|||||||
|
Directors |
||||||||||||
|
Joel Latham |
278,731 |
115,000 |
— |
24,627 |
217,712 |
636,070 |
||||||
|
Troy Valentine |
54,000 |
— |
127,500 |
5,130 |
— |
186,630 |
||||||
|
Dr. Sud Agarwal |
48,000 |
— |
90,000 |
4,560 |
454,987 |
597,547 |
||||||
|
Peter Widdows |
48,000 |
— |
— |
4,560 |
— |
52,560 |
||||||
|
Other Key Management Personnel |
||||||||||||
|
Madhukar Bhalla |
60,000 |
— |
— |
— |
— |
— |
||||||
|
428,731 |
115,000 |
217,500 |
38,877 |
672,699 |
1,472,807 |
|||||||
Service Agreements
The following members of key personnel have service agreements as at June 30, 2021 as follows:
|
Joel Latham |
Managing Director and Chief Executive Officer |
|
|
Agreement commenced: |
July 1, 2020 |
|
|
Details |
This employment agreement has no fixed term. Each party can terminate at will by giving three months’ notice. However, if the termination is for cause, no notice is required. |
|
|
Base salary including superannuation |
A$260,000 per year, including a vehicle allowance. |
|
|
Madhukar Bhalla |
Chief Financial Officer and Company Secretary |
|
|
Agreement commenced: |
June 28, 2021 |
|
|
Details |
This service agreement has no fixed term. This service agreement can be terminated by either party at will by giving 1-month notice. |
|
|
Base salary including superannuation |
A$60,000 per year for services as Chief Financial Officer and Company Secretary. |
|
|
Dr. Sud Agarwal |
Chief Medical Officer |
|
|
Agreement commenced: |
July 23, 2019 |
|
|
Details |
This service agreement has a fixed term of one year and it automatically renews if the parties do not terminate it. Dr. Sud Agarwal can terminate with 90 days notice. Either party can terminate the contract without notice in the case of material breach or insolvency. |
|
|
Base salary including superannuation |
A$90,000 per year for services as Chief Medical Officer. |
Employee Share Option Plan and Performance Rights Plan
The Company does not currently have any Employee Share Option Plan or Performance Rights Plan. In the event that the directors determined that such plans were necessary, the Company would seek shareholder approval for any such plan prior to their use.
88
Over the past three years, the Company has issued options or performance rights to directors or management as part of their remuneration or as performance incentives. No options or performance rights were granted to directors and officers during fiscal year 2021. All of these issues have been approved by shareholders prior to their issuance. Details of these issues are below:
|
Recipient |
Quantity |
Type |
Shareholder approval date |
|||
|
Joel Latham |
8,000,000 |
Performance Rights |
20 November 2018 |
|||
|
Troy Valentine |
2,500,000 |
Performance Rights |
20 November 2018 |
|||
|
Peter Widdows |
2,500,000 |
Performance Rights |
20 November 2018 |
|||
|
Madhukar Bhalla |
— |
— |
— |
|||
|
Dr. Sud Agarwal |
32,303,593 |
Performance Rights |
26 June 2020 |
|||
|
Dr. Sud Agarwal |
200,000,000 |
Options |
26 June 2020 |
|||
|
Joel Latham |
4,500,000 |
Options |
26 June 2020 |
Ordinary Share holdings
As at June 30, 2021, the numbers of shares held by our directors and officers were as follows.
|
2021 |
Balance at |
Received on |
Received |
Other |
Balance at |
||||||
|
Ordinary shares |
|
||||||||||
|
Joel Latham |
11,829,129 |
— |
2,000,000 |
4,119,285 |
(1) |
17,948,414 |
|||||
|
Troy Valentine |
20,234,248 |
— |
6,500,000 |
— |
|
26,734,248 |
|||||
|
Dr. Sud Agarwal |
36,000,000 |
30,303,593 |
— |
— |
|
66,303,593 |
|||||
|
Peter Widdows |
12,615,790 |
— |
3,300,000 |
— |
|
15,915,790 |
|||||
|
Madhukar Bhalla |
— |
— |
— |
— |
|
— |
|||||
|
Total ordinary shares |
80,679,167 |
30,303,593 |
11,800,000 |
4,119,285 |
|
126,902,045 |
|||||
____________
(1) Removal from voluntary escrow.
Options holdings
As at June 30, 2021, the numbers of options held by our directors and officers were as follows. Each options grants the right to receive one fully paid ordinary share in Incannex.
|
2021 |
Balance at |
Exercise price |
Expiration date |
Changes |
Balance at |
||||||
|
Options |
|
||||||||||
|
Joel Latham |
4,237,500 |
|
A$ 0.04 |
September 30, 2020 |
— |
4,237,500 |
|||||
|
Joel Latham |
— |
$ |
0.05 |
June 30, 2025 |
750,000 |
750,000 |
|||||
|
Joel Latham |
— |
$ |
0.05 |
June 30, 2026 |
750,000 |
750,000 |
|||||
|
Joel Latham |
— |
$ |
0.05 |
June 30, 2027 |
750,000 |
750,000 |
|||||
|
Joel Latham |
— |
$ |
0.08 |
September 30, 2021 |
200,000 |
200,000 |
|||||
|
Troy Valentine |
$ |
0.08 |
September 30, 2021 |
7,116,950 |
7,116,950 |
||||||
|
Dr. Sud Agarwal(1) (2) |
200,000,000 |
|
A$ 0.20 |
September 30, 2021 |
— |
200,000,000 |
|||||
|
Peter Widdows(3) |
657,895 |
|
A$ 0.08 |
September 30, 2021 |
— |
657,895 |
|||||
|
Madhukar Bhalla |
— |
|
— |
— |
— |
— |
|||||
|
Total options |
204,895,395 |
|
9,566,950 |
214,462,345 |
|||||||
____________
(1) Granted to Dr. Sud Agarwal directly.
(2) 88,000,000 options granted to Cannvalate Pty Ltd, in which Dr. Sud Agarwal is a director and significant shareholder, expired during fiscal year 2021. In particular, (i) 14,000,000 options, exercise price A$0.06 per option, expired on December 31, 2020, (ii) 16,000,000 options, exercise price $0.08 per option, expired on December 31, 2020,
89
(iii) 18,000,000 options, exercise price A$0.10 per option, expired on December 31, 2020, (iv) 20,000,000 options, exercise price A$0.12 per option, expired on December 31, 2020, and (v) 20,000,000 options, exercise price A$0.14 per option, expired on December 31, 2020.
(3) 3,300,000 options, exercise price A$0.04 per option, expired on September 30, 2020.
Performance rights
As at June 30, 2021, the numbers of performance rights held by our directors and officers were as follows. Each performance right grants the right to receive one fully paid ordinary share in the Company.
|
2021 |
Balance at |
Granted/(Expired) |
Converted to |
Balance at |
||||||
|
Performance rights |
|
|
||||||||
|
Joel Latham |
5,000,000 |
(5,000,000 |
) |
— |
|
— |
||||
|
Troy Valentine |
1,500,000 |
(1,500,000 |
) |
— |
|
— |
||||
|
Dr. Sud Agarwal |
32,303,593 |
(2,000,000 |
) |
(30,303,593 |
) |
— |
||||
|
Peter Widdows |
1,500,000 |
(1,500,000 |
) |
— |
|
— |
||||
|
Madhukar Bhalla |
— |
— |
|
— |
|
— |
||||
|
Total performance rights |
40,303,593 |
(10,000,000 |
) |
(30,303,593 |
) |
— |
||||
Board Practices
Introduction
Our Board of Directors is elected by and accountable to our shareholders. It currently consists of four directors, including three non-executive directors, of which one is the non-executive Chairman of our Board of Directors. The Chairman of our Board of Directors is responsible for the management of the Board of Directors and its functions.
Election of Directors
Directors are elected at our annual general meeting of shareholders. Under our Constitution, a director, other than a managing director, must not hold office for more than three years or beyond the third annual general meeting following his appointment (whichever is the longer period) without submitting himself for re-election. Our Board of Directors has the power to appoint any person to be a director, either to fill a vacancy or as an additional director (provided that the total number of directors does not exceed the maximum allowed by law), and any director so appointed may hold office only until the next annual general meeting (“AGM”) when he or she shall be eligible for election.
The appointment and expiration dates of each director in office on June 30, 2021, is as follows:
|
Name |
Position |
Year first appointed |
Current term expires |
|||
|
Joel Latham |
Managing Director and CEO |
2018 |
—(1) |
|||
|
Troy Valentine |
Chairman |
2019 |
2022(2) |
|||
|
Dr. Sud Agarwal |
Chief Medical Officer and Director |
2019 |
2021(2) |
|||
|
Peter Widdows |
Director |
2020 |
2023(2) |
____________
(1) According to our Constitution, a Managing Director’s appointment is not subject to expiration.
(2) Term expires on the date of the AGM for that year.
90
Corporate Governance
ASX Corporate Governance Principles
In Australia, there are no defined corporate governance structures and practices that must be observed by a company listed on the ASX, except that entities of a certain size are required to have audit and remuneration committees and, in some instances, trading policies for key management personnel. Instead, the ASX Corporate Governance Council has published the Corporate Governance Principles and Recommendations, which contains what are called the Recommendations which articulate eight core principles which are intended to provide a reference point for companies about their corporate governance structures and practices. Under ASX Listing Rule 4.10.3, companies are required to attach a copy of the Company’s corporate governance statement (which has been approved by the Board) and provide a statement in their annual report to shareholders disclosing the extent to which they have followed the Recommendations in the reporting period and where they have not followed all the Recommendations, identify the Recommendations that have not been followed, and the reasons for not following them and what (if any) alternative governance practices it adopted in lieu of the recommendations during that period. It is not mandatory to follow the Recommendations. As compliance with the Recommendations would entail excessive costs to us, and in light of our current size, we do not follow the Recommendations because the costs of doing so would outweigh the benefits.
Non-Executive and Independent Directors
Australian law does not require a company to appoint a certain number of independent directors to its board of directors or audit committee. However, under the Corporate Governance Principles and Recommendations, the ASX recommends, but does not require, that an ASX-listed company have a majority of independent directors on its board of directors. Our Board of Directors has determined that each of Troy Valentine and Peter Widdows qualifies as an independent director under the requirements of the ASX.
Our Board of Directors does not have regularly scheduled meetings at which only independent directors are present. The Board of Directors does meet regularly and independent directors are expected to attend all such meetings.
Committees of the Board of Directors
Audit Committee. Nasdaq Marketplace Rules require us to establish an audit committee comprised of at least three members, each of whom is financially literate and satisfies the respective “independence” requirements of the SEC and Nasdaq and one of whom has accounting or related financial management expertise at senior levels within a company.
Our Audit Committee assists our Board of Directors in overseeing the accounting and financial reporting processes of our company and audits of our financial statements, including the integrity of our financial statements, compliance with legal and regulatory requirements, our independent public accountants’ qualifications and independence, and independent public accountants, and such other duties as may be directed by our Board of Directors. The Audit Committee is also required to assess risk management.
Our Audit Committee currently consists of two board members, Peter Widdows and Troy Valentine. Each of Troy Valentine and Peter Widdows satisfies the “independence” requirements of the U.S. Securities and Exchange Commission and Nasdaq Marketplace Rules. As permitted by Nasdaq Marketplace Rules, we will appoint a third independent board member to the audit committee within 1 year of listing on Nasdaq. The audit committee meets at least two times per year.
91
Corporate Governance Requirements under Nasdaq listing rules.
As we are incorporated in Australia, we are allowed to follow Australian “home country” corporate governance practices in lieu of the otherwise applicable Nasdaq corporate governance standards, as long as we disclose each requirement of Rule 5600 that we do not follow and describe the home country practice we follow in lieu of the relevant Nasdaq corporate governance standards. We intend to take all actions necessary to maintain compliance with applicable corporate governance requirements under the rules adopted by the SEC and listing standards of Nasdaq. We follow Australian corporate governance practices in lieu of the corporate governance requirements of the Nasdaq Marketplace Rules in respect of:
• Nasdaq requirement under Rule 5605(d) that a compensation committee be constituted — The ASX Listing Rules do not have an express requirement that each issuer listed on ASX have a compensation committee. We expect to rely on an exemption from the requirement to constitute a compensation committee under the Nasdaq listing rules and we seek to claim such exemption.
• Nasdaq requirement under Rule 5605(e) that a nominations committee be constituted — The ASX Listing Rules do not have an express requirement that each issuer listed on ASX have a nominations committee. We expect to rely on an exemption from the requirement to constitute a nominations committee under the Nasdaq listing rules and we seek to claim such exemption.
• Nasdaq requirement under Rule 5620(c) that a quorum consist of holders of 33 1/3% of the outstanding ordinary shares — The ASX Listing Rules do not have an express requirement that each issuer listed on ASX have a quorum of any particular number of the outstanding ordinary shares, but instead allow a listed issuer to establish its own quorum requirements. Our quorum is currently two persons who are entitled to vote. We believe this quorum requirement is consistent with the requirements of the ASX and is appropriate and typical of generally accepted business practices in Australia.
• Nasdaq requirements under Rules 5605(b)(1) and (2) relating to director independence, including the requirements that a majority of the board of directors must be comprised of independent directors and that independent directors must have regularly scheduled meetings at which only independent directors are present — The Nasdaq and ASX definitions of what constitute an independent director are not identical and the requirements relating to the roles and obligations of independent directors are not identical. The ASX, unlike Nasdaq, permits an issuer to establish its own materiality threshold for determining whether a transaction between a director and an issuer affects the director’s status as independent and it does not require that a majority of the issuer’s board of directors be independent, as long as the issuer publicly discloses this fact. In addition, the ASX does not require that the independent directors have regularly scheduled meeting at which only independent directors are present. We believe that our Board composition is consistent with the requirements of the ASX and that it is appropriate and typical of generally accepted business practices in Australia.
• The requirement that our independent directors meet regularly in executive sessions under Nasdaq Listing Rules. The ASX Listing Rules and the Corporations Act do not require the independent directors of an Australian company to have such executive sessions.
• The Nasdaq requirements under Rules 5605(d) that compensation of an issuer’s officers must be determined, or recommended to the Board for determination, either by a majority of the independent directors, or a compensation committee comprised solely of independent directors, and that director nominees must either be selected, or recommended for the Board’s selection, either by a majority of the independent directors, or a nominations committee comprised solely of independent directors. The Nasdaq compensation committee requirements are not identical to the ASX remuneration and nomination committee requirements. Issuers listed on the ASX are recommended under applicable listing standards to establish a remuneration committee consisting of a majority of independent directors and an independent chairperson, or publicly disclose that it has not done so. We do not have a compensation committee.
92
• The requirement prescribed by Nasdaq Listing Rules that issuers obtain shareholder approval prior to the issuance of securities in connection with certain acquisitions, private placements of securities, or the establishment or amendment of certain share option, purchase or other compensation plans. Applicable Australian law and the ASX Listing Rules differ from Nasdaq requirements, with the ASX Listing Rules providing generally for prior shareholder approval in numerous circumstances, including (i) issuance of equity securities exceeding 15% (or 25% under certain circumstances) of our issued share capital in any 12-month period (but, in determining the 15% limit, securities issued under an exception to the rule or with shareholder approval are not counted), (ii) issuance of equity securities to related parties (as defined in the ASX Listing Rules) and (iii) issuances of securities to directors or their associates under an employee incentive plan.
Indemnification of Directors and Officers
Our Constitution provides that, we may indemnify a person who is, or has been, a director or an officer of our company, to the full extent permissible by law, out of our property against any liability incurred by such person as a director or an officer in defending proceedings, whether civil or criminal, and whatever their outcome.
In addition, our Constitution provides that to the extent permitted by law, we may pay, or agree to pay, a premium in respect of a contract insuring a person who is or has been a director or an officer of our company or one of our subsidiaries against any liability:
• incurred by the person in his or her capacity as a director or an officer of our company or a subsidiary of our company, and
• for costs and expenses incurred by that person in defending proceedings relating to that person acting as a director or an officer of Incannex, whether civil or criminal, and whatever their outcome.
We maintain a directors’ and officers’ liability insurance policy. We have established a policy for the indemnification of our directors and officers against certain liabilities incurred as a director or officer, including costs and expenses associated in successfully defending legal proceedings
Employees
As of June 30, 2021, we had 4 employees. Of these employees, 3 were employed in research and development and 1 in general management and administration. All the employees were located in Australia. As at the end of fiscal year 2020, we had 4 employees.
Each of our full-time employees has entered into an agreement with an unlimited term. We may only terminate the employment of any of our employees in accordance with the relevant employee’s contract of employment.
Our standard contract of employment for full time provides that we can terminate the employment of an employee without notice for serious misconduct or with between one to six months’ notice without cause (as set out in the relevant employee’s contract of employment).
93
Share Ownership
Ownership of Senior Management and Directors
The following table sets forth certain information as of June 30, 2021 regarding the ownership of our ordinary shares by each of our directors and senior management and by all of our directors and senior management as a group. The percentages shown are based on 1,068,411,224 ordinary shares issued and outstanding as of June 30, 2021.
|
Name |
Number of Ordinary Shares |
Percentage of Ownership |
|||
|
Joel Latham |
17,948,414 |
1.68 |
% |
||
|
Troy Valentine(2) |
26,734,248 |
2.50 |
% |
||
|
Dr. Sud Agarwal(1) |
34,303,593 |
3.21 |
% |
||
|
Peter Widdows |
15,915,790 |
1.49 |
% |
||
|
Madhukar Bhalla |
— |
— |
|
||
|
All directors and executive officers as a group (5 persons) – |
94,902,045 |
8.88 |
% |
||
____________
(1) Dr. Sud Agarwal also owns 200,000,000 options to purchase ordinary shares. In addition, Dr. Sud Agarwal owns approximately 30% of the ordinary shares in Cannvalate, which owns 32,000,000 ordinary shares of Incannex. Dr. Sud Agarwal, as major shareholder and director of Cannvalate, may be deemed to have voting and dispositive power with respect to the ordinary shares in Incannex held by Cannvalate. Please see “Principal Shareholders” to see beneficial interest including Cannvalate’s interest in Incannex.
(2) Troy Valentine is a director, and owns a 50% equity interest in, Alignment Capital Pty Ltd. Thus, Troy Valentine is deemed to beneficially own 13,194,248 ordinary shares that Alignment Capital Pty Ltd owns in Incannex.
Code of Conduct
We have adopted a Code of Conduct applicable to all of our directors, officers and employees. Our Code of Conduct is available on our website at www.incannex.com.au. We post on our website all disclosures that are required by law or the listing standards of Nasdaq concerning any amendments to, or waivers from, any provision of the Code of Conduct. The reference to our website address does not constitute incorporation by reference of the information contained at or available through our website, and you should not consider it to be a part of, this prospectus.
94
Certain Relationships and Related Party Transactions
The following is a description of our related party transactions since July 1, 2018 and we note that all of them were negotiated at arm’s length.
In fiscal years 2021, 2020 and 2019, respectively, the Company paid A$97,976, A$145,200 and A$115,864 in fees to Alignment Capital Pty Ltd (“Alignment”), an entity controlled by our Chairman Troy Valentine, as consideration for its services as lead manager.
In June 2019, the Company borrowed A$15,000 from Joel Latham, our Chief Executive Officer, and A$50,000 from Alignment to secure funds to continue the Company’s operations while in the process of completing a capital raising. These funds were advanced with no interest or security element. These amounts were fully repaid by June 30, 2019.
In March 2019, we entered into a distribution agreement with Cannvalate Pty Ltd, a company in which Dr. Sud Agarwal is a director and major shareholder. Under the terms of the agreement, we had the right to distribute cannabinoid oil products in Australia through Cannvalate’s network. This agreement was terminated on June 30, 2021.
95
The following table presents the beneficial ownership of our ordinary shares based on 1,068,411,224 ordinary shares outstanding at June 30, 2021 by each person known by us to be the beneficial owner of more than 5% of our ordinary shares.
We have determined beneficial ownership in accordance with the rules and regulations of the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Except as indicated by the footnotes below, we believe, based on information furnished to us, that the persons and entities named in the table below have sole voting and sole investment power with respect to all shares that they beneficially own.
Applicable percentage ownership before the offering is based on 1,068,411,224 ordinary shares outstanding as of June 30, 2021. Applicable percentage ownership after the offering is based on ordinary shares outstanding immediately after the closing of this offering (after giving effect to the sale and issuance of ADSs representing ordinary shares at an ADS-to-ordinary share ratio of 1-to-50), assuming no exercise by the underwriter of its option to purchase additional ADSs. In computing the number of shares beneficially owned by a person or entity and the percentage ownership of such person or entity, we deemed to be outstanding all shares subject to options and warrants held by the person or entity that are currently exercisable, or exercisable within 60 days of June 30, 2021. However, except as described above, we did not deem such shares outstanding for the purpose of computing the percentage ownership of any other person or entity.
|
Ordinary |
Ordinary |
||||||||
|
Shareholder |
Number |
Percentage |
Number |
Percentage |
|||||
|
Dr. Sud Agarwal(1) |
266,303,593 |
19.96 |
% |
266,303,593 |
|||||
____________
(1) Includes 34,303,593 ordinary shares, 200,000,000 options to purchase ordinary shares and 32,000,000 ordinary shares owned by Cannvalate, in which Dr. Sud Agarwal owns approximately 30% and is Chairman and, as such, may be deemed to have voting and dispositive power with respect to the ordinary shares in Incannex held by Cannvalate.
As of June 30, 2021, there were 7,449 holders of record of our ordinary shares, of which none had registered addresses in the United States. These numbers are not representative of the number of beneficial holders of our shares nor are they representative of where such beneficial holders reside, as many of these ordinary shares were held of record by brokers or other nominees.
To our knowledge, we are not directly or indirectly controlled by another corporation, by any foreign government or by any other natural or legal person severally or jointly. There are no agreements known to us, the operation of which may at a subsequent date result in a change in control of Incannex. All shareholders have the same voting rights.
96
General
As of June 30, 2021, we had (i) 1,068,411,224 fully paid ordinary shares outstanding, and (ii) 326,437,328 options outstanding at a weighted average exercise price of A$0.1655.
Memorandum and Articles of Association
General
Our constituent document is a Constitution. The Constitution is subject to the terms of the Listing Rules of ASX Limited and the Corporations Act 2001. The Constitution may be amended or repealed and replaced by special resolution of shareholders, which is a resolution of which notice has been given and that has been passed by at least 75% of the votes cast by shareholders entitled to vote on the resolution.
Purposes and Objects
As a public company we have all the rights, powers and privileges of a natural person. Our Constitution does not provide for or prescribe any specific objects or purposes.
The Powers of the Directors
Under the provision of our Constitution our directors may exercise all the powers of our company except any powers that the Corporations Act or the constitution attributes to Incannex.
Interested Directors
According to our constitution, if a Director discloses his or her in accordance with the Corporations Act, the director may (i) contract or make an arrangement with the Company, or a related body corporate of the Company or a body corporate in which the Company is interested, in any matter in any capacity, (ii) be counted in a quorum for a meeting of Directors considering the contract or arrangement, (iii) vote on whether the Company enters into the contract or arrangement, and on any matter that relates to the contract or arrangement, (iv) sign on behalf of the Company, or witness the affixing of the common seal of the Company to, any document in respect of the contract or arrangement, (v) retain the benefits under the contract or arrangement.
Unless a relevant exception applies, the Corporations Act requires our directors to provide disclosure of certain interests or conflicts of interests and prohibits directors from voting on matters in which they have a material personal interest and from being present at the meeting while the matter is being considered. In addition, the Corporations Act and the ASX Listing Rules require shareholder approval of any provision of related party benefits to our directors.
Directors’ compensation
Our non-executive directors are paid remuneration for their services as directors which is determined in a general meeting of shareholders. The aggregate, fixed sum for directors’ remuneration is to be divided among the directors in such proportion as the directors themselves agree and in accordance with our Constitution. Our executive directors are paid remuneration for their services as directors which is determined by all directors.
Fees payable to our non-executive directors must be by way of a fixed sum and not by way of a commission on or a percentage of profits or operating revenue. Remuneration paid to our executive directors must also not include a commission or percentage of operating revenue.
Pursuant to our Constitution, any director who performs services that in the opinion of our board of directors, are outside the scope of the ordinary duties of a director may be paid extra remuneration, which is determined by our board of directors.
97
In addition to other remuneration provided in our Constitution, all of our directors are entitled to be paid by us for reasonable travel accommodation and other expenses incurred by the directors in attending general meetings, board meetings, committee meetings or otherwise in connection with our business.
In addition, in accordance with our Constitution, a director may be paid a retirement benefit as determined by our board of directors subject to the limits set out in the Corporations Act and the ASX Listing Rules which broadly restrict our ability to pay our officers a termination benefit in the event of a change of control of the Company or our subsidiaries as well as impose requirements for shareholder approval to be obtained to pay certain retirement benefits to our officers.
Borrowing powers exercisable by Directors
Pursuant to our Constitution, the management and control of our business affairs are vested in our board of directors. Thus, our board of directors has the power to raise or borrow money, and charge any of our property or business or any uncalled capital, and may issue debentures or give any other security for any of our debts, liabilities or obligations or of any other person, in each case, in the manner and on terms it deems fit.
Retirement of Directors
Pursuant to our Constitution and the ASX Listing Rules, each director, other than the managing director, must not hold office for more than three years or beyond the third annual general meeting following his or her appointment (whichever is longer). Further, at least one director is required to retire by rotation at each annual general meeting (such director being the director who has been longest in office since their last election). Directors who retire by rotation are eligible for a re-election to the board of directors unless disqualified from acting as a director under the Corporations Act or our Constitution.
Rights Attached to Our Ordinary Shares
The concept of authorized share capital no longer exists in Australia and as a result, our authorized share capital is unlimited. All our outstanding ordinary shares are validly issued, fully paid and non-assessable. The rights attached to our ordinary shares are as follows:
Dividend Rights.
The directors may declare that a dividend be paid to the members according to the shareholders’ pro rata shareholdings and the directors may fix the amount, the time for payment and the method of payment. No dividend is payable except in accordance with the Corporations Act as amended from time to time and no dividend carries interest as against the Company.
Voting Rights.
Holders of ordinary shares have one vote for each ordinary share held on all matters submitted to a vote of shareholders. Such voting rights may be affected by the grant of any special voting rights to the holders of a class of shares with preferential rights that may be authorized in the future.
The quorum required for an ordinary meeting of shareholders consists of at least two shareholders present in person, or by proxy, attorney or representative appointed pursuant to our Constitution. A meeting adjourned for lack of a quorum generally is adjourned to the same day in the following week at the same time and place. At the reconvened meeting, the required quorum consists of any two members present in person, or by proxy, attorney or representative appointed pursuant to our Constitution. The meeting is dissolved if a quorum is not present within 30 minutes from the time appointed for the meeting.
An ordinary resolution, such as a resolution for the declaration of dividends, requires approval by the holders of a majority of the voting rights represented at the meeting, in person, by proxy, or by written ballot and voting thereon. Under our Constitution, the Corporations Act and the ASX Listing Rules, certain matters must be passed by way of a special resolution. A special resolution must be passed by at least 75% of the votes cast by shareholders entitled to vote on the resolution and who vote at the meeting in person. Matters which are not required to be passed by special resolution are required to be passed by ordinary resolution.
98
Rights in Our Profits.
Our shareholders have the right to share in our profits distributed as a dividend and any other permitted distribution.
Rights in the Event of Liquidation.
In the event of our liquidation, after satisfaction of liabilities to creditors, our assets will be distributed to the holders of ordinary shares in proportion to the capital at the commencement of the liquidation paid up or which ought to have been paid up on the shares held by them respectively. This right may be affected by the grant of preferential dividend or distribution rights to the holders of a class of shares with preferential rights, such as the right in winding up to payment in cash of the amount then paid up on the share, and any arrears of dividend in respect of that share, in priority to any other class of shares.
Directors may make calls
Our Constitution provides that subject to the terms on which the shares have been issued directors may make calls on a shareholder for amounts unpaid on shares held by that shareholder, other than monies payable at fixed times under the conditions of allotment.
Changing Rights Attached to Shares
According to our Constitution, the rights attached to any class of shares, unless otherwise provided by the terms of the class, may be varied with either the written consent of the holders of not less than 75% of the issued shares of that class or the sanction of a special resolution passed at a separate general meeting of the shares of that class.
Annual and Extraordinary Meetings
Our directors must convene an annual meeting of shareholders at least once every calendar year. Notice of at least 28 days prior to the date of the meeting is required. A general meeting may be convened by any director, or shareholders in compliance with the Corporations Act.
Limitations on the Rights to Own Securities in Our Company
Subject to certain limitations on the percentage of shares a person may hold in our company, neither our Constitution nor the laws of the Commonwealth of Australia restrict in any way the ownership or voting of shares in our company.
Changes in Our Capital
Pursuant to the Listing Rules, our directors may in their discretion issue securities to persons who are not related parties of our company, without the approval of shareholders, if such issue, when aggregated with securities issued by our company during the previous 12-month period would be an amount that would not exceed 15% of our issued capital at the commencement of the 12 month period. Other allotments of securities require approval by an ordinary resolution of shareholders.
99
Description of American Depositary Shares
American Depositary Shares
Deutsche Bank Trust Company Americas, as depositary, will register and deliver the ADSs. Each ADS will represent ownership of 50 ordinary shares, deposited with National Nominees Limited, as custodian for the depositary. Each ADS will also represent ownership of any other securities, cash or other property which may be held by the depositary. The depositary’s corporate trust office at which the ADSs will be administered is located at 60 Wall Street, New York, NY 10005, USA. The principal executive office of the depositary is located at 60 Wall Street, New York, NY 10005, USA.
The Direct Registration System, or DRS, is a system administered by The Depository Trust Company, or DTC, pursuant to which the depositary may register the ownership of uncertificated ADSs, which ownership shall be evidenced by periodic statements issued by the depositary to the ADS holders entitled thereto.
We will not treat ADS holders as our shareholders and accordingly, you, as an ADS holder, will not have shareholder rights. Australian law governs shareholder rights. The depositary will be the holder of the ordinary shares underlying your ADSs. As a holder of ADSs, you will have ADS holder rights. A deposit agreement among us, the depositary and you, as an ADS holder, and the beneficial owners of ADSs sets out ADS holder rights as well as the rights and obligations of the depositary. The laws of the State of New York govern the deposit agreement and the ADSs. See “— Jurisdiction and Arbitration.”
The following is a summary of the material provisions of the deposit agreement. For more complete information, you should read the entire deposit agreement and the form of American Depositary Receipt. For directions on how to obtain copies of those documents, see “Where You Can Find Additional Information.”
Holding the ADSs
How will you hold your ADSs?
You may hold ADSs either (i) directly (a) by having an American Depositary Receipt, or ADR, which is a certificate evidencing a specific number of ADSs, registered in your name, or (b) by holding ADSs in DRS, or (ii) indirectly through your broker or other financial institution. If you hold ADSs directly, you are an ADS holder. This description assumes you hold your ADSs directly. ADSs will be issued through DRS, unless you specifically request certificated ADRs. If you hold the ADSs indirectly, you must rely on the procedures of your broker or other financial institution to assert the rights of ADS holders described in this section. You should consult with your broker or financial institution to find out what those procedures are.
Dividends and Other Distributions
How will you receive dividends and other distributions on the shares?
The depositary has agreed to pay to you the cash dividends or other distributions it or the custodian receives on ordinary shares or other deposited securities, after deducting its fees and expenses. You will receive these distributions in proportion to the number of ordinary shares your ADSs represent as of the record date (which will be as close as practicable to the record date for our ordinary shares) set by the depositary with respect to the ADSs.
• Cash. The depositary will convert or cause to be converted any cash dividend or other cash distribution we pay on the ordinary shares or any net proceeds from the sale of any ordinary shares, rights, securities or other entitlements under the terms of the deposit agreement into U.S. dollars if it can do so on a practicable basis and can transfer the U.S. dollars to the United States and will distribute promptly the amount thus received. If the depositary shall determine in its judgment that such conversions or transfers are not practical or lawful or if any government approval or license is needed and cannot be obtained at a reasonable cost within a reasonable period or otherwise sought, the deposit agreement allows the depositary to distribute the foreign currency only to those ADS holders to whom it is possible to do so. It will hold or cause the custodian to hold the foreign currency it cannot convert for the account of the ADS holders who have not been paid and such funds will be held for the respective accounts of the ADS holders. It will not invest the foreign currency and it will not be liable for any interest for the respective accounts of the ADS holders.
100
Before making a distribution, any taxes or other governmental charges, together with fees and expenses of the depositary, that must be paid, will be deducted. See “Taxation.” It will distribute only whole U.S. dollars and cents and will round down fractional cents to the nearest whole cent. If the exchange rates fluctuate during a time when the depositary cannot convert the foreign currency, you may lose some or all of the value of the distribution.
• Shares. For any ordinary shares we distribute as a dividend or free distribution, either (1) the depositary will distribute additional ADSs representing such ordinary shares or (2) existing ADSs as of the applicable record date will represent rights and interests in the additional ordinary shares distributed, to the extent reasonably practicable and permissible under law, in either case, net of applicable fees, charges and expenses incurred by the depositary and taxes and/or other governmental charges. The depositary will only distribute whole ADSs. It will try to sell ordinary shares which would require it to deliver a fractional ADS and distribute the net proceeds in the same way as it does with cash. The depositary may sell a portion of the distributed ordinary shares sufficient to pay its fees and expenses, and any taxes and governmental charges, in connection with that distribution.
• Elective Distributions in Cash or Shares. If we offer holders of our ordinary shares the option to receive dividends in either cash or shares, the depositary, after consultation with us and having received timely notice as described in the deposit agreement of such elective distribution by us, has discretion to determine to what extent such elective distribution will be made available to you as a holder of the ADSs. We must timely first instruct the depositary to make such elective distribution available to you and furnish it with satisfactory evidence that it is legal to do so. The depositary could decide it is not legal or reasonably practicable to make such elective distribution available to you. In such case, the depositary shall, on the basis of the same determination as is made in respect of the ordinary shares for which no election is made, distribute either cash in the same way as it does in a cash distribution, or additional ADSs representing ordinary shares in the same way as it does in a share distribution. The depositary is not obligated to make available to you a method to receive the elective dividend in shares rather than in ADSs. There can be no assurance that you will be given the opportunity to receive elective distributions on the same terms and conditions as the holders of ordinary shares.
• Rights to Purchase Additional Shares. If we offer holders of our ordinary shares any rights to subscribe for additional shares, the depositary shall having received timely notice as described in the deposit agreement of such distribution by us, consult with us, and we must determine whether it is lawful and reasonably practicable to make these rights available to you. We must first instruct the depositary to make such rights available to you and furnish the depositary with satisfactory evidence that it is legal to do so. If the depositary decides it is not legal or reasonably practicable to make the rights available but that it is lawful and reasonably practicable to sell the rights, the depositary will endeavor to sell the rights and in a riskless principal capacity or otherwise, at such place and upon such terms (including public or private sale) as it may deem proper distribute the net proceeds in the same way as it does with cash.
The depositary will allow rights that are not distributed or sold to lapse. In that case, you will receive no value for them.
If the depositary makes rights available to you, it will establish procedures to distribute such rights and enable you to exercise the rights upon your payment of applicable fees, charges and expenses incurred by the depositary and taxes and/or other governmental charges. The Depositary shall not be obliged to make available to you a method to exercise such rights to subscribe for ordinary shares (rather than ADSs).
U.S. securities laws may restrict transfers and cancellation of the ADSs represented by shares purchased upon exercise of rights. For example, you may not be able to trade these ADSs freely in the United States. In this case, the depositary may deliver restricted depositary shares that have the same terms as the ADSs described in this section except for changes needed to put the necessary restrictions in place.
There can be no assurance that you will be given the opportunity to exercise rights on the same terms and conditions as the holders of ordinary shares or be able to exercise such rights.
101
• Other Distributions. Subject to receipt of timely notice, as described in the deposit agreement, from us with the request to make any such distribution available to you, and provided the depositary has determined such distribution is lawful and reasonably practicable and feasible and in accordance with the terms of the deposit agreement, the depositary will distribute to you anything else we distribute on deposited securities by any means it may deem practicable, upon your payment of applicable fees, charges and expenses incurred by the depositary and taxes and/or other governmental charges. If any of the conditions above are not met, the depositary will endeavor to sell, or cause to be sold, what we distributed and distribute the net proceeds in the same way as it does with cash; or, if it is unable to sell such property, the depositary may dispose of such property in any way it deems reasonably practicable under the circumstances for nominal or no consideration, such that you may have no rights to or arising from such property.
The depositary is not responsible if it decides that it is unlawful or impractical to make a distribution available to any ADS holders. We have no obligation to register ADSs, shares, rights or other securities under the Securities Act. We also have no obligation to take any other action to permit the distribution of ADSs, shares, rights or anything else to ADS holders. This means that you may not receive the distributions we make on our shares or any value for them if we and/or the depositary determines that it is illegal or not practicable for us or the depositary to make them available to you.
Deposit, Withdrawal and Cancellation
How are ADSs issued?
The depositary will deliver ADSs if you or your broker deposit ordinary shares or evidence of rights to receive ordinary shares with the custodian. Upon payment of its fees and expenses and of any taxes or charges, such as stamp taxes or stock transfer taxes or fees, the depositary will register the appropriate number of ADSs in the names you request and will deliver the ADSs to or upon the order of the person or persons entitled thereto.
Except for ordinary shares deposited by us in connection with this offering, no shares will be accepted for deposit prior to the date of this prospectus.
How do ADS holders cancel an American Depositary Share?
You may turn in your ADSs at the depositary’s corporate trust office or by providing appropriate instructions to your broker. Upon payment of its fees and expenses and of any taxes or charges, such as stamp taxes or stock transfer taxes or fees, the depositary will deliver the ordinary shares and any other deposited securities underlying the ADSs to you or a person you designate at the office of the custodian. Or, at your request, risk and expense, the depositary will deliver the deposited securities at its corporate trust office, to the extent permitted by law.
How do ADS holders interchange between Certificated ADSs and Uncertificated ADSs?
You may surrender your ADR to the depositary for the purpose of exchanging your ADR for uncertificated ADSs. The depositary will cancel that ADR and will send you a statement confirming that you are the owner of uncertificated ADSs. Alternatively, upon receipt by the depositary of a proper instruction from a holder of uncertificated ADSs requesting the exchange of uncertificated ADSs for certificated ADSs, the depositary will execute and deliver to you an ADR evidencing those ADSs.
Voting Rights
How do you vote?
You may instruct the depositary to vote the ordinary shares or other deposited securities underlying your ADSs at any meeting at which you are entitled to vote pursuant to any applicable law, the provisions of our constitution, and the provisions of or governing the deposited securities. Otherwise, you could exercise your right to vote directly if you withdraw the ordinary shares. However, you may not know about the meeting sufficiently enough in advance to withdraw the ordinary shares.
102
If we ask for your instructions and upon timely notice from us by regular, ordinary mail delivery, or by electronic transmission, as described in the deposit agreement, the depositary will notify you of the upcoming meeting at which you are entitled to vote pursuant to any applicable law, the provisions of our constitution, and the provisions of or governing the deposited securities, and arrange to deliver our voting materials to you. The materials will include or reproduce (a) such notice of meeting or solicitation of consents or proxies; (b) a statement that the ADS holders at the close of business on the ADS record date will be entitled, subject to any applicable law, the provisions of our constitution, and the provisions of or governing the deposited securities, to instruct the depositary as to the exercise of the voting rights, if any, pertaining to the ordinary shares or other deposited securities represented by such holder’s ADSs; and (c) a brief statement as to the manner in which such instructions may be given to the depositary. Voting instructions may be given only in respect of a number of ADSs representing an integral number of ordinary shares or other deposited securities. For instructions to be valid, the depositary must receive them in writing on or before the date specified. The depositary will try, as far as practical, subject to applicable law and the provisions of our constitution, to vote or to have its agents vote the ordinary shares or other deposited securities (in person or by proxy) as you instruct.
We cannot assure you that you will receive the voting materials in time to ensure that you can instruct the depositary to vote the ordinary shares underlying your ADSs. In addition, there can be no assurance that ADS holders and beneficial owners generally, or any holder or beneficial owner in particular, will be given the opportunity to vote or cause the custodian to vote on the same terms and conditions as the holders of our ordinary shares.
The depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that you may not be able to exercise your right to vote and you may have no recourse if the ordinary shares underlying your ADSs are not voted as you requested.
In order to give you a reasonable opportunity to instruct the depositary as to the exercise of voting rights relating to deposited securities, if we request the depositary to act, we will give the depositary notice of any such meeting and details concerning the matters to be voted at least 28 Business Days in advance of the meeting date.
Compliance with Regulations
Information Requests
Each ADS holder and beneficial owner shall (a) provide such information as we or the depositary may request pursuant to law, including, without limitation, relevant Australian law, any applicable law of the United States of America, our constitution, any resolutions of our Board of Directors adopted pursuant to such constitution, the requirements of any markets or exchanges upon which the ordinary shares, ADSs or ADRs are listed or traded, or to any requirements of any electronic book-entry system by which the ADSs or ADRs may be transferred, regarding the capacity in which they own or owned ADRs, the identity of any other persons then or previously interested in such ADRs and the nature of such interest, and any other applicable matters, and (b) be bound by and subject to applicable provisions of the laws of the Australia, our constitution, and the requirements of any markets or exchanges upon which the ADSs, ADRs or ordinary shares are listed or traded, or pursuant to any requirements of any electronic book-entry system by which the ADSs, ADRs or ordinary shares may be transferred, to the same extent as if such ADS holder or beneficial owner held ordinary shares directly, in each case irrespective of whether or not they are ADS holders or beneficial owners at the time such request is made.
Disclosure of Interests
Each ADS holder and beneficial owner shall comply with our requests pursuant to Australian law, the rules and requirements of the Nasdaq and any other stock exchange on which the ordinary shares are, or will be, registered, traded or listed or our constitution, which requests are made to provide information, inter alia, as to the capacity in which such ADS holder or beneficial owner owns ADS and regarding the identity of any other person interested in such ADS and the nature of such interest and various other matters, whether or not they are ADS holders or beneficial owners at the time of such requests.
103
Fees and Expenses
As an ADS holder, you will be required to pay the following service fees to the depositary bank and certain taxes and governmental charges (in addition to any applicable fees, expenses, taxes and other governmental charges payable on the deposited securities represented by any of your ADSs):
|
Service |
Fees |
|
|
To any person to which ADSs are issued or to any person to which a distribution is made in respect of ADS distributions pursuant to stock dividends or other free distributions of stock, bonus distributions, stock splits or other distributions (except where converted to cash) |
Up to US$0.05 per ADS issued |
|
|
Cancellation of ADSs, including the case of termination of the deposit agreement |
Up to US$0.05 per ADS cancelled |
|
|
Distribution of cash dividends |
Up to US$0.05 per ADS held |
|
|
Distribution of cash entitlements (other than cash dividends) and/or cash proceeds from the sale of rights, securities and other entitlements |
Up to US$0.05 per ADS held |
|
|
Distribution of ADSs pursuant to exercise of rights. |
Up to US$0.05 per ADS held |
|
|
Depositary services |
Up to US$0.04 per ADS held on the applicable record date(s) established by the depositary bank |
As an ADS holder, you will also be responsible for paying certain fees and expenses incurred by the depositary bank and certain taxes and governmental charges (in addition to any applicable fees, expenses, taxes and other governmental charges payable on the deposited securities represented by any of your ADSs) such as:
• Taxes (including applicable interest and penalties) and other governmental charges;
• Fees for the transfer and registration of ordinary shares charged by the registrar and transfer agent for the ordinary shares in the Australian (i.e., upon deposit and withdrawal of ordinary shares).
• Expenses incurred for converting foreign currency into U.S. dollars.
• Expenses for cable, telex and fax transmissions and for delivery of securities.
• Taxes and duties upon the transfer of securities, including any applicable stamp duties, any stock transfer charges or withholding taxes (i.e., when ordinary shares are deposited or withdrawn from deposit).
• Fees and expenses incurred in connection with the delivery or servicing of ordinary shares on deposit.
• Fees and expenses incurred in connection with complying with exchange control regulations and other regulatory requirements applicable to ordinary shares, deposited securities, ADSs and ADRs.
• Any applicable fees and penalties thereon.
The depositary fees payable upon the issuance and cancellation of ADSs are typically paid to the depositary bank by the brokers (on behalf of their clients) receiving the newly issued ADSs from the depositary bank and by the brokers (on behalf of their clients) delivering the ADSs to the depositary bank for cancellation. The brokers in turn charge these fees to their clients. Depositary fees payable in connection with distributions of cash or securities to ADS holders and the depositary services fee are charged by the depositary bank to the holders of record of ADSs as of the applicable ADS record date.
The depositary fees payable for cash distributions are generally deducted from the cash being distributed or by selling a portion of distributable property to pay the fees. In the case of distributions other than cash (i.e., share dividends, rights), the depositary bank charges the applicable fee to the ADS record date holders concurrent with the distribution. In the case of ADSs registered in the name of the investor (whether certificated or uncertificated in direct registration), the depositary bank sends invoices to the applicable record date ADS holders. In the case of
104
ADSs held in brokerage and custodian accounts (via DTC), the depositary bank generally collects its fees through the systems provided by DTC (whose nominee is the registered holder of the ADSs held in DTC) from the brokers and custodians holding ADSs in their DTC accounts. The brokers and custodians who hold their clients’ ADSs in DTC accounts in turn charge their clients’ accounts the amount of the fees paid to the depositary banks.
In the event of refusal to pay the depositary fees, the depositary bank may, under the terms of the deposit agreement, refuse the requested service until payment is received or may set off the amount of the depositary fees from any distribution to be made to the ADS holder.
The depositary may make payments to us or reimburse us for certain costs and expenses, by making available a portion of the ADS fees collected in respect of the ADR program or otherwise, upon such terms and conditions as we and the depositary bank agree from time to time.
Payment of Taxes
You will be responsible for any taxes or other governmental charges payable, or which become payable, on your ADSs or on the deposited securities represented by any of your ADSs. The depositary may refuse to register or transfer your ADSs or allow you to withdraw the deposited securities represented by your ADSs until such taxes or other charges are paid. It may apply payments owed to you or sell deposited securities represented by your ADSs to pay any taxes owed and you will remain liable for any deficiency. If the depositary sells deposited securities, it will, if appropriate, reduce the number of ADSs to reflect the sale and pay to you any net proceeds, or send to you any property, remaining after it has paid the taxes. You agree to indemnify us, the depositary, the custodian and each of our and their respective agents, directors, employees and affiliates for, and hold each of them harmless from, any claims with respect to taxes (including applicable interest and penalties thereon) arising from any refund of taxes, reduced rate of withholding at source or other tax benefit obtained for you. Your obligations under this paragraph shall survive any transfer of ADRs, any surrender of ADRs and withdrawal of deposited securities or the termination of the deposit agreement.
Reclassifications, Recapitalizations and Mergers
|
If we: |
Then: |
|
|
Change the nominal or par value of our ordinary shares |
The shares or other securities received by the depositary will become deposited securities. |
|
|
Reclassify, split up or consolidate any of the deposited securities |
Each ADS will automatically represent its equal share of the new deposited securities. |
|
|
Distribute securities on the ordinary shares that are not distributed to you, or recapitalize, reorganize, merge, liquidate, sell all or substantially all of our assets, or take any similar action |
The depositary may distribute some or all of the cash, shares or other securities it received. It may also deliver new ADSs or ask you to surrender your outstanding ADRs in exchange for new ADRs identifying the new deposited securities. |
Amendment and Termination
How may the deposit agreement be amended?
We may agree with the depositary to amend the deposit agreement and the form of ADR without your consent for any reason. If an amendment adds or increases fees or charges, except for taxes and other governmental charges or expenses of the depositary for registration fees, facsimile costs, delivery charges or similar items, including expenses incurred in connection with foreign exchange control regulations and other charges specifically payable by ADS holders under the deposit agreement, or materially prejudices a substantial existing right of ADS holders, it will not become effective for outstanding ADSs until 30 days after the depositary notifies ADS holders of the amendment. At the time an amendment becomes effective, you are considered, by continuing to hold your ADSs, to agree to the amendment and to be bound by the ADRs and the deposit agreement as amended. If any new laws are adopted which would require the deposit agreement to be amended in order to comply therewith, we and the depositary may amend the deposit agreement in accordance with such laws and such amendment may become effective before notice thereof is given to ADS holders.
105
How may the deposit agreement be terminated?
The depositary will terminate the deposit agreement if we ask it to do so, in which case the depositary will give notice to you at least 90 days prior to termination. The depositary may also terminate the deposit agreement if the depositary has told us that it would like to resign, or if we have removed the depositary, and in either case we have not appointed a new depositary within 90 days. In either such case, the depositary must notify you at least 30 days before termination.
After termination, the depositary and its agents will do the following under the deposit agreement but nothing else: collect distributions on the deposited securities, sell rights and other property and deliver ordinary shares and other deposited securities upon cancellation of ADSs after payment of any fees, charges, taxes or other governmental charges. Six months or more after the date of termination, the depositary may sell any remaining deposited securities by public or private sale. After that, the depositary will hold the money it received on the sale, as well as any other cash it is holding under the deposit agreement, for the pro rata benefit of the ADS holders that have not surrendered their ADSs. It will not invest the money and has no liability for interest. After such sale, the depositary’s only obligations will be to account for the money and other cash. After termination, we shall be discharged from all obligations under the deposit agreement except for our obligations to the depositary thereunder.
Books of Depositary
The depositary will maintain ADS holder records at its depositary office. You may inspect such records at such office during regular business hours but solely for the purpose of communicating with other holders in the interest of business matters relating to the Company, the ADRs and the deposit agreement.
The depositary will maintain facilities in the Borough of Manhattan, The City of New York to record and process the issuance, cancellation, combination, split-up and transfer of ADRs.
These facilities may be closed at any time or from time to time when such action is deemed necessary or advisable by the depositary in connection with the performance of its duties under the deposit agreement or at our reasonable written request.
Limitations on Obligations and Liability
Limits on our Obligations and the Obligations of the Depositary and the Custodian; Limits on Liability to Holders of ADSs
The deposit agreement expressly limits our obligations and the obligations of the depositary and the custodian. It also limits our liability and the liability of the depositary. The depositary and the custodian:
• are only obligated to take the actions specifically set forth in the deposit agreement without gross negligence or willful misconduct;
• are not liable if any of us or our respective controlling persons or agents are prevented or forbidden from, or subjected to any civil or criminal penalty or restraint on account of, or delayed in, doing or performing any act or thing required by the terms of the deposit agreement and any ADR, by reason of any provision of any present or future law or regulation of the United States or any state thereof, the Commonwealth of Australia or any other country, or of any other governmental authority or regulatory authority or stock exchange, or on account of the possible criminal or civil penalties or restraint, or by reason of any provision, present or future, of our memorandum and articles of association or any provision of or governing any deposited securities, or by reason of any act of God or war or other circumstances beyond its control (including, without limitation, nationalization, expropriation, currency restrictions, work stoppage, strikes, civil unrest, revolutions, rebellions, explosions and computer failure);
• are not liable by reason of any exercise of, or failure to exercise, any discretion provided for in the deposit agreement or in our memorandum and articles of association or provisions of or governing deposited securities;
106
• are not liable for any action or inaction of the depositary, the custodian or us or their or our respective controlling persons or agents in reliance upon the advice of or information from legal counsel, any person presenting ordinary shares for deposit or any other person believed by it in good faith to be competent to give such advice or information;
• are not liable for the inability of any holder of ADSs to benefit from any distribution on deposited securities that is not made available to holders of ADSs under the terms of the deposit agreement;
• are not liable for any special, consequential, indirect or punitive damages for any breach of the terms of the deposit agreement, or otherwise;
• may rely upon any documents we believe in good faith to be genuine and to have been signed or presented by the proper party;
• disclaim any liability for any action or inaction or inaction of any of us or our respective controlling persons or agents in reliance upon the advice of or information from legal counsel, accountants, any person presenting ordinary shares for deposit, holders and beneficial owners (or authorized representatives) of ADSs, or any person believed in good faith to be competent to give such advice or information; and
• disclaim any liability for inability of any holder to benefit from any distribution, offering, right or other benefit made available to holders of deposited securities but not made available to holders of ADS.
The depositary and any of its agents also disclaim any liability (i) for any failure to carry out any instructions to vote, the manner in which any vote is cast or the effect of any vote or failure to determine that any distribution or action may be lawful or reasonably practicable or for allowing any rights to lapse in accordance with the provisions of the deposit agreement, (ii) the failure or timeliness of any notice from us, the content of any information submitted to it by us for distribution to you or for any inaccuracy of any translation thereof, (iii) any investment risk associated with the acquisition of an interest in the deposited securities, the validity or worth of the deposited securities, the credit-worthiness of any third party, (iv) for any tax consequences that may result from ownership of ADSs, ordinary shares or deposited securities, or (v) for any acts or omissions made by a successor depositary, provided that in connection with the issue out of which such potential liability arises the depositary performed its obligations without gross negligence or wilful misconduct while it acted as depositary.
In the deposit agreement, we agree to indemnify the depositary under certain circumstances.
Jurisdiction and Arbitration
The laws of the State of New York govern the deposit agreement and the ADSs and we have agreed with the depositary that the federal or state courts in the City of New York shall have exclusive jurisdiction to hear and determine any dispute arising from or in connection with the deposit agreement and that the depositary will have the right to refer any claim or dispute arising from the relationship created by the deposit agreement to arbitration in accordance with the Commercial Arbitration Rules of the American Arbitration Association. The arbitration provisions of the deposit agreement do not preclude you from pursuing claims under the Securities Act or the Exchange Act in federal or state courts.
Jury Trial Waiver
The deposit agreement provides that each party to the deposit agreement (including each holder, beneficial owner and holder of interests in the ADRs) irrevocably waives, to the fullest extent permitted by applicable law, any right it may have to a trial by jury in any lawsuit or proceeding against us or the depositary arising out of or relating to our shares, the ADSs or the deposit agreement, including any claim under the U.S. federal securities laws. If we or the depositary opposed a jury trial demand based on the waiver, the court would determine whether the waiver was enforceable based on the facts and circumstances of that case in accordance with the applicable law.
107
Requirements for Depositary Actions
Before the depositary will issue, deliver or register a transfer of an ADS, split-up, subdivide or combine ADSs, make a distribution on an ADS, or permit withdrawal of ordinary shares, the depositary may require:
• payment of stock transfer or other taxes or other governmental charges and transfer or registration fees charged by third parties for the transfer of any ordinary shares or other deposited securities and payment of the applicable fees, expenses and charges of the depositary;
• satisfactory proof of the identity and genuineness of any signature or any other matters contemplated in the deposit agreement; and
• compliance with (A) any laws or governmental regulations relating to the execution and delivery of ADRs or ADSs or to the withdrawal or delivery of deposited securities and (B) such reasonable regulations and procedures as the depositary may establish, from time to time, consistent with the deposit agreement and applicable laws, including presentation of transfer documents.
The depositary may refuse to issue and deliver ADSs or register transfers of ADSs generally when the register of the depositary or our transfer books are closed or at any time if the depositary or we determine that it is necessary or advisable to do so.
Your Right to Receive the Shares Underlying Your ADSs
You have the right to cancel your ADSs and withdraw the underlying ordinary shares at any time except:
• when temporary delays arise because: (1) the depositary has closed its transfer books or we have closed our transfer books; (2) the transfer of ordinary shares is blocked to permit voting at a shareholders’ meeting; or (3) we are paying a dividend on our ordinary shares;
• when you owe money to pay fees, taxes and similar charges;
• when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities, or other circumstances specifically contemplated by Section I.A.(l) of the General Instructions to Form F-6 (as such General Instructions may be amended from time to time); or
• for any other reason if the depositary or we determine, in good faith, that it is necessary or advisable to prohibit withdrawals.
The depositary shall not knowingly accept for deposit under the deposit agreement any ordinary shares or other deposited securities required to be registered under the provisions of the Securities Act, unless a registration statement is in effect as to such ordinary shares.
This right of withdrawal may not be limited by any other provision of the deposit agreement.
Direct Registration System
In the deposit agreement, all parties to the deposit agreement acknowledge that the DRS and Profile Modification System, or Profile, will apply to uncertificated ADSs upon acceptance thereof to DRS by DTC. DRS is the system administered by DTC pursuant to which the depositary may register the ownership of uncertificated ADSs, which ownership shall be evidenced by periodic statements issued by the depositary to the ADS holders entitled thereto. Profile is a required feature of DRS which allows a DTC participant, claiming to act on behalf of an ADS holder, to direct the depositary to register a transfer of those ADSs to DTC or its nominee and to deliver those ADSs to the DTC account of that DTC participant without receipt by the depositary of prior authorization from the ADS holder to register such transfer.
108
Shares Eligible for Future Sale
Upon completion of this offering, we will have ADSs outstanding, representing ordinary shares, or approximately % of our outstanding ordinary shares, assuming the underwriter does not exercise its option to purchase additional ADSs. All of the ADSs sold in this offering will be freely transferable by persons other than our “affiliates” without restriction or further registration under the Securities Act. Sales of substantial amounts of the ADSs in the public market could adversely affect prevailing market prices of the ADSs. Prior to this offering, there has been no public market for the ADSs, and while we have applied to have the ADSs listed on the Nasdaq we cannot assure you that a regular trading market will develop in the ADSs.
Lock-up Agreements
Our directors and officers have agreed, subject to some exceptions, not to transfer or dispose of, directly or indirectly, any of our ordinary shares, in the form of ADSs or otherwise, or any securities convertible into or exchangeable or exercisable for our ordinary shares, in the form of ADSs or otherwise, for a period of 180 days after the date of this prospectus. After the expiration of the 180-day period, the ordinary shares or ADSs held by our directors, executive officers and our existing shareholders may be sold subject to the restrictions under Rule 144 under the Securities Act or by means of registered public offerings.
Rule 144
All of our ordinary shares outstanding prior to this offering are “restricted shares” as defined in Rule 144 under the Securities Act and may be sold publicly in the United States only if they are subject to an effective registration statement under the Securities Act or pursuant to an exemption from the registration requirements. Under Rule 144 as currently in effect, a person who has beneficially owned our restricted shares for at least six months is generally entitled to sell the restricted securities without registration under the Securities Act beginning 90 days after the date of this prospectus, subject to certain additional restrictions.
Our affiliates may sell within any three-month period a number of restricted shares that does not exceed the greater of the following:
• 1% of the then outstanding ordinary shares of the same class, in the form of ADSs or otherwise, which will equal approximately ordinary shares immediately after this offering, assuming the underwriter does not exercise its option to purchase additional ADSs; or
• the average weekly trading volume of our ordinary shares in the form of ADSs or otherwise on the Nasdaq during the four calendar weeks preceding the date on which notice of the sale is filed with the SEC.
Affiliates who sell restricted securities under Rule 144 may not solicit orders or arrange for the solicitation of orders, and they are also subject to notice requirements and the availability of current public information about us.
Persons who are not our affiliates are only subject to one of these additional restrictions, the requirement of the availability of current public information about us, and this additional restriction does not apply if they have beneficially owned our restricted shares for more than one year.
Rule 701
In general, under Rule 701 of the Securities Act as currently in effect, each of our employees, consultants or advisors who purchases our ordinary shares from us in connection with a compensatory shares or option plan or other written agreement relating to compensation is eligible to resell such ordinary shares 90 days after we became a reporting company under the Exchange Act in reliance on Rule 144, but without compliance with some of the restrictions, including the holding period, contained in Rule 144.
109
Material United States Federal Income and Australian Tax Considerations
The following summary of the material Australian and U.S. federal income tax consequences of an investment in the ADSs or ordinary shares is based upon laws and relevant interpretations thereof in effect as of the date of this prospectus, all of which are subject to change, possibly with retroactive effect. This summary does not deal with all possible tax consequences relating to an investment in the ADSs or ordinary shares, such as the tax consequences under U.S. state, local and other tax laws other than U.S. federal income tax laws and certain Australian tax laws.
Holders of our ADSs should consult their own tax advisors as to the United States, Australian or other tax consequences of the purchase, ownership and disposition of ADSs, including, in particular, the effect of any foreign, state or local taxes.
Australian Taxation
In this section we discuss the material Australian tax considerations that apply to non-Australian tax residents with respect to the acquisition, ownership and disposal of the absolute beneficial ownership of ADSs, which are evidenced by ADSs. This discussion is based upon existing Australian tax law as of the date of this Registration Statement, which is subject to change, possibly retrospectively. This discussion does not address all aspects of Australian income tax law which may be important to particular investors in light of their individual investment circumstances, such as ADSs or shares held by investors subject to special tax rules (for example, financial institutions, insurance companies or tax exempt organizations). In addition, this summary does not discuss any foreign or state tax considerations, other than stamp duty. Prospective investors are urged to consult their tax advisors regarding the Australian and foreign income and other tax considerations of the purchase, ownership and disposition of the ADSs or shares.
Nature of ADSs for Australian Taxation Purposes
Holders of our ADSs are treated as the owners of the underlying ordinary shares for Australian income tax and capital gains tax purposes. Therefore, dividends paid on the underlying ordinary shares will be treated for Australian tax purposes as if they were paid directly to the owners of ADSs, and the disposal of ADSs will be treated for Australian tax purposes as the disposal of the underlying ordinary shares. In the following analysis we discuss the application of the Australian income tax and capital gains tax rules to non-Australian resident holders of ADSs.
Taxation of Dividends
Australia operates a dividend imputation system under which dividends may be declared to be “franked” to the extent they are paid out of company profits that have been subject to income tax. Fully franked dividends are not subject to dividend withholding tax. Dividends that are not franked or are partly franked and are paid to non-Australian resident shareholders are subject to dividend withholding tax, but only to the extent the dividends are not franked.
Dividends paid to a non-resident shareholder are subject to withholding tax (a) except to the extent they have been franked and (b) at 30%, unless the shareholder is a resident of a country with which Australia has a double taxation agreement.
In accordance with the provisions of the Double Taxation Convention between Australia and the United States, the maximum rate of Australian tax on any unfranked portion of a dividend to which a resident of the United States is beneficially entitled is 15%, where the U.S. resident holds less than 10% of the voting rights in our company, or 5% where the U.S. resident holds 10% or more of the voting rights in our company. Special rules apply to Regulated Investment Companies and Real Estate Investment Trusts that hold shares and receive dividends. The Double Taxation Convention between Australia and the United States does not apply to limit the tax rate on dividends where the ADSs are effectively connected to a permanent establishment or a fixed base carried on by the owner of the ADSs in Australia through which the shareholder carries on business or provides independent personal services, respectively.
110
Tax on Sales or other Dispositions of Shares — Capital Gains Tax
Australian capital gains derived by non-Australian residents in respect of the disposal of capital assets that are not taxable Australian property will be disregarded. Non-Australian resident shareholders will not be subject to Australian capital gains tax on the capital gain made on a disposal of our shares, unless they, together with associates, hold 10% or more of our issued capital, tested either at the time of disposal or over any continuous 12- month period in the 24 months prior to disposal, and the value of our shares at the time of disposal is principally attributable to Australian real property assets.
Australian capital gains tax applies to net capital gains at a taxpayer’s marginal tax rate but for certain shareholders a discount capital gain may apply if the shares have been held for 12 months or more. For individuals, this discount is 50%. Net capital gains are calculated after reduction for capital losses (including certain prior year capital losses), which may only be offset against capital gains.
Tax on Sales or other Dispositions of Shares — Shareholders Holding Shares on Revenue Account
Some non-Australian resident shareholders may hold shares on revenue rather than on capital account, for example, share traders. These shareholders may have the gains made on the sale or other disposal of the shares included in their assessable income under the ordinary income provisions of the income tax law, if the gains are sourced in Australia.
Non-Australian resident shareholders assessable under these ordinary income provisions in respect of gains made on shares held on revenue account would be assessed for such gains at the Australian tax rates for non-Australian residents, which start at a marginal rate of 32.5% for non-Australian resident individuals. Some relief from the Australian income tax may be available to such non-Australian resident shareholders under the Double Taxation Convention between the United States and Australia, for example, because the shareholder does not have a permanent establishment in Australia.
To the extent an amount would be included in a non-Australian resident shareholder’s assessable income under both the capital gains tax provisions and the ordinary income provisions, the capital gain amount would generally be reduced, so that the shareholder would not be subject to double tax on any part of the income gain or capital gain.
Dual Residency
If a shareholder were a resident of both Australia and the United States under those countries’ domestic taxation laws, that shareholder may be subject to tax as an Australian resident. If, however, the shareholder is determined to be a U.S. resident for the purposes of the Double Taxation Convention between the United States and Australia, the Australian tax applicable would be limited by the Double Taxation Convention. Shareholders should obtain specialist taxation advice in these circumstances.
Stamp Duty
A transfer of shares of a company listed on the Australian Securities Exchange is not subject to Australian stamp duty.
Australian Death Duty
Australia does not have estate or death duties. No capital gains tax liability is realized upon the inheritance of a deceased person’s shares. The disposal of inherited shares by beneficiaries, may, however, give rise to a capital gains tax liability.
Goods and Services Tax
The issue or transfer of shares will not incur Australian goods and services tax.
U.S. Taxation
The following is a summary of material U.S. federal income tax consequences that generally apply to U.S. Holders (as defined below) who hold ADSs as capital assets within the meaning of Section 1221 of the Internal Revenue Code of 1986, as amended (the “Code”). This summary is based on the Code, its legislative history, final,
111
temporary and proposed United States Treasury regulations promulgated thereunder, published rulings and court decisions, and the bilateral income tax convention between Australia and the United States (the “Treaty”), all as in effect on the date hereof and all of which are subject to change, or changes in interpretation, either prospectively or retroactively. This discussion does not address all of the tax consequences relating to the purchase, ownership, and disposition of ADSs and does not take into account U.S. Holders who may be subject to special rules, including: financial institutions, insurance companies, , tax-exempt organizations, real estate investment trusts, regulated investment companies, grantor trusts, non-resident aliens of the United States or taxpayers whose functional currency is not the U.S. dollar, persons who hold the ADSs through partnerships or other pass-through entities, persons who acquired their ADSs through the exercise or cancellation of any employee share options or otherwise as compensation for their services, investors that actually or constructively own 10% or more of our shares, dealers or traders in securities or currencies, certain former citizens or long-term residents of the United States, dual resident corporations, persons that generally mark their securities to market for United States federal income tax purposes, persons who are residents of Australia for Australian income tax purposes, and investors holding ADSs as part of a straddle or appreciated financial position or as part of a hedging or conversion transaction. This summary does not address the Medicare tax imposed on certain investment income, any state, local and foreign tax considerations or any U.S. federal estate, gift or alternative minimum tax considerations relevant to the purchase, ownership and disposition of our ADSs. In addition, this discussion is based in part upon representations of the depositary and the assumption that each obligation in the deposit agreement and any related agreements will be performed according to its terms.
If a partnership or an entity or arrangement treated as a partnership for U.S. federal income tax purposes owns ADSs, the U.S. federal income tax treatment of its partners will generally depend upon the status of the partner and the activities of the partnership. A partnership should consult its tax advisors regarding the U.S. federal income tax consequences applicable to it and its partners of the purchase, ownership and disposition of ADSs.
For purposes of this summary, the term “U.S. Holder” means a beneficial owner of ADSs that is for U.S. federal income tax purposes: an individual who is a citizen or resident of the United States; a corporation that is created or organized in or under the laws of the United States or any political subdivision thereof; an estate whose income is subject to U.S. federal income tax regardless of its source; or a trust if (a) a court within the United States is able to exercise primary supervision over administration of the trust, and one or more U.S. persons have the authority to control all substantial decisions of the trust or (b) it has a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person.
Distributions
For U.S. federal income tax purposes, a U.S. Holder of ADSs will be treated as owning the ordinary shares underlying the ADSs. Subject to the passive foreign investment company rules discussed below, the gross amount of any distribution received by a U.S. Holder with respect to our ordinary shares or ADSs, including the amount of any Australian taxes withheld therefrom, will be included in gross income as a dividend to the extent the distribution is paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). Distributions in excess of our earnings and profits will be treated first as a non-taxable return of capital to the extent of a U.S. Holder’s tax basis in the ADSs and thereafter will be treated as gain from the sale or exchange of the ADSs. We have not maintained and do not plan to maintain calculations of earnings and profits for U.S. federal income tax purposes. As a result, a U.S. Holder may need to include the entire amount of any such distribution in income as a dividend. Dividends will not, however, be eligible for the “dividends received deduction” generally allowed to corporate shareholders with respect to dividends received from U.S. corporations.
The U.S. dollar value of any distribution on the ADSs made in Australian dollars generally should be calculated by reference to the spot exchange rate between the U.S. dollar and the Australian dollar in effect on the date the distribution is actually or constructively received by the U.S. Holder regardless of whether the Australian dollars so received are in fact converted into U.S. dollars. A U.S. Holder who receives payment in Australian dollars and converts those Australian dollars into U.S. dollars at an exchange rate other than the rate in effect on such day may have a foreign currency exchange gain or loss, which would generally be treated as ordinary income or loss from sources within the United States for U.S. foreign tax credit purposes.
Subject to complex limitations and certain holding period requirements, a U.S. Holder may elect to claim a credit for Australian tax withheld from distributions against its U.S. federal income tax liability. The limitations set out in the Code include computational rules under which foreign tax credits allowable with respect to specific
112
classes of income cannot exceed the U.S. federal income taxes otherwise payable with respect to each such class of income. Dividends generally will be treated as foreign-source passive category income for U.S. foreign tax credit purposes or in the case of certain U.S. Holders as foreign source “general category” income. A U.S. Holder that does not elect to claim a U.S. foreign tax credit may instead claim a deduction for Australian tax withheld.
Subject to certain limitations, dividends received by a non-corporate U.S. Holder are subject to tax at a reduced maximum tax rate of 20 percent if the dividends are “qualified dividends”. Dividends are qualified dividends if: (a)(i) the issuer is entitled to benefits under the Treaty or (ii) the shares are readily tradable on an established securities market in the United States and (b) certain other requirements are met. We believe that we are entitled to benefits under the Treaty and that the ADSs currently are readily tradable on an established securities market in the United States. However, no assurance can be given that the ADSs will remain readily tradable. Further, the reduced rate does not apply to dividends if we are a PFIC in the year prior to or the year in which the dividend is paid.
The U.S. Treasury has expressed concerns that intermediaries in the chain of ownership between the holder of an ADS and the issuer of the security underlying the ADS may be taking actions that are inconsistent with the claiming of foreign tax credits for U.S. Holders of ADSs. Such actions would also be inconsistent with the claiming of the reduced rate of tax, described above, applicable to dividends received by certain non-corporate holders. Accordingly, the analysis of the creditability of Australian taxes and the availability of the reduced tax rate for dividends received by certain non-corporate holders, each described above, could be affected by actions taken by intermediaries in the chain of ownership between the holder of an ADS and our Company.
Disposition of ADSs
If you sell or otherwise dispose of ADSs, you will recognize gain or loss for U.S. federal income tax purposes in an amount equal to the difference between the U.S. dollar value of the amount realized on the sale or other disposition and your adjusted tax basis in the ADSs. Subject to the passive foreign investment company rules discussed below, such gain or loss generally will be capital gain or loss and will be long-term capital gain or loss if you have held the ADSs for more than one year at the time of the sale or other disposition. In general, any gain that you recognize on the sale or other disposition of ADSs will be gain from U.S. sources for purposes of the foreign tax credit limitation; losses will generally be allocated against U.S. source income. The deduction of capital losses is subject to certain limitations under the Code.
In the case of a cash-basis U.S. Holder who receives Australian dollars in connection with the sale or other disposition of ADSs, the amount realized will be calculated based on the U.S. dollar value of the Australian dollars received as determined by reference to the spot rate in effect on the settlement date of such exchange. A U.S. Holder who receives payment in Australian dollars and converts Australian dollars into U.S. dollars at a conversion rate other than the rate in effect on the settlement date may have foreign currency exchange gain or loss that would be treated as ordinary income or loss from sources within the United States for U.S. foreign tax credit purposes.
An accrual-basis U.S. Holder may elect the same treatment required of cash-basis taxpayers with respect to a sale or disposition of ADSs, provided that the election is applied consistently from year to year. Such election may not be changed without the consent of the Internal Revenue Service (“IRS”). In the event that an accrual-basis U.S. Holder does not elect to be treated as a cash-basis taxpayer (pursuant to the Treasury regulations applicable to foreign currency transactions), such U.S. Holder may have foreign currency gain or loss for U.S. federal income tax purposes because of differences between the U.S. dollar value of the currency received prevailing on the trade date and the settlement date. Any such currency gain or loss would be treated as ordinary income or loss from sources within the United States for U.S. foreign tax credit purposes. However, if foreign currency is converted into U.S. dollars on the date received by the U.S. Holder, a cash-basis or electing accrual-basis U.S. Holder should not recognize any gain or loss on such conversion.
Passive Foreign Investment Companies
There is a risk that we may be a passive foreign investment company(“PFIC”), for U.S. federal income tax purposes. Our treatment as a PFIC could result in a reduction in the after-tax return to the U.S. Holders of our ADSs and may cause a reduction in the value of such securities.
113
For U.S. federal income tax purposes, we will be classified as a PFIC for any taxable year in which (i) 75% or more of our gross income is passive income, or (ii) at least 50% of the average value of all of our assets for the taxable year produce or are held for the production of passive income. For this purpose, cash is considered to be an asset which produces passive income. Passive income for these purposes generally includes dividends, interest, royalties, rents, annuities and the excess of gains over losses from the disposition of assets which produce passive income. In making a PFIC determination, we will be treated as owning our proportionate share of the assets and earning our proportionate share of the income of any other corporation in which we own, directly or indirectly, 25% or more (by value) of the share capital. Based on the composition of our assets and income, we believe that we should not be treated as a PFIC for U.S. federal income tax purposes with respect to fiscal year 2020. However, the determination of PFIC status is a factual determination that must be made annually at the close of each taxable year and, therefore, there can be no certainty as to our status in this regard until the close of the current or any future taxable year. Changes in the nature of our income or assets or a decrease in the trading price of our ADSs may cause us to be considered a PFIC in the current or any subsequent year. If we were a PFIC in any year during a U.S. Holder’s holding period for our ADSs, we would ordinarily continue to be treated as a PFIC for each subsequent year during which the U.S. Holder owned the ADSs.
Under the default PFIC “excess distribution” regime, if we are a PFIC in any taxable year during which a U.S. Holder owns ADSs, such U.S. Holder could be liable for additional taxes and interest charges upon (i) certain distributions by us (generally any distribution paid during a taxable year that is greater than 125 percent of the average annual distributions paid in the three preceding taxable years, or, if shorter, the U.S. Holder’s holding period for the ADSs), and (ii) any gain realized on a sale, exchange or other disposition, including a pledge, of the ADSs, whether or not we continue to be a PFIC for the year of the disposition. In these circumstances, the tax will generally be determined by allocating such distributions or gain ratably over the U.S. Holder’s holding period for the ADSs. The amount allocated to the current taxable year and any year prior to the first taxable year in which we are a PFIC will be taxed as ordinary income (rather than capital gain) earned in the current taxable year. The amount allocated to other taxable years will be taxed at the highest applicable marginal rates for the year and an interest charge at the rate applicable to underpayments of tax will also be imposed on the amount of taxes allocated to such other taxable years.
An indirect shareholder may be taxed on a distribution paid to the direct owner of a PFIC and on a disposition of the share indirectly owned. Indirect shareholders are strongly urged to consult their tax advisors regarding the application of these rules.
If we are a PFIC and subsequently cease to be a PFIC in a future year, a U.S. Holder may avoid the continued application of the tax treatment described above by electing to be treated as if it sold its ADSs on the last day of the last taxable year in which we were a PFIC. Any gain would generally be recognized and subject to tax under the excess distribution regime described above. Loss would not be recognized. A U.S. Holder’s basis in its ADSs would be increased by the amount of gain, if any, recognized on the deemed sale. A U.S. Holder would be required to treat its holding period for its ADSs as beginning on the day following the last day of the last taxable year in which we were a PFIC.
If the ADSs are considered “marketable stock” and if a U.S. Holder properly elects to “mark-to-market” its ADSs in a timely fashion, the U.S. Holder would not be subject to tax under the excess distribution regime described above. Instead, the U.S. Holder would generally include in income any excess of the fair market value of the ADSs at the close of each tax year over the adjusted tax basis of the ADSs. If the fair market value of the ADSs had depreciated below the adjusted basis at the close of the tax year, the U.S. Holder would be entitled to deduct the excess of the adjusted basis of the ADSs over their fair market value at that time. However, such deductions generally would be limited to the net mark-to-market gains, if any, the U.S. Holder included in income with respect to such ADSs in prior years. Income recognized and deductions allowed under the mark-to-market provisions, as well as any gain or loss on the disposition of ADSs with respect to which the mark-to-market election is made, is treated as ordinary income or loss (except that loss is treated as capital loss to the extent the loss exceeds the net mark-to-market gains, if any, that a U.S. Holder included in income with respect to such ordinary shares in prior years). However, gain or loss from the disposition of ADSs (as to which a “mark-to-market” election was properly made) in a year in which we are no longer a PFIC, will be capital gain or loss. Our ordinary shares or ADSs will be “marketable” stock as long as they remain regularly traded on a national securities exchange, such as the Nasdaq, or a foreign securities exchange regulated by a governmental authority of the country in which the market is located
114
and which meets certain requirements, including that the rules of the exchange effectively promote active trading of listed stocks. If such stock is traded on such a qualified exchange or other market, such stock generally will be “regularly traded” for any calendar year during which such stock is traded, other than in de minimis quantities, on at least 15 days during each calendar quarter, but no assurances can be given in this regard. Our ordinary shares are traded on the ASX, which may qualify as an eligible foreign securities exchange for this purpose. Because a mark-to-market election cannot be made for any lower-tier PFICs that we may own, a U.S. Holder may continue to be subject to the PFIC rules with respect to such holder’s indirect interest in any investments held by us that are treated as an equity interest in a PFIC for U.S. federal income tax purposes, including shares in any of our subsidiaries that are treated as PFICs.
A U.S. Holder of ADSs should not be able to avoid the tax consequences described above by electing to treat us as a qualified electing fund. In general, a qualified electing fund is, with respect to a U.S. person, a PFIC if the U.S. person has elected to include its proportionate share of a company’s ordinary earnings and net capital gains in U.S. income on an annual basis. A qualified electing fund election can only be made with respect to us if we provide U.S. Holders with certain information on an annual basis and we do not intend to prepare the information that U.S. Holders would need to make the qualified electing fund election.
Backup Withholding and Information Reporting
Payments in respect of ADSs may be subject to information reporting to the IRS and to U.S. backup withholding tax (at a rate of 24% under current law). Backup withholding will not apply, however, if a U.S. Holder (i) is a corporation, (ii) satisfies an applicable exemption, or (iii) furnishes correct taxpayer identification.
Backup withholding is not an additional tax. Amounts withheld under the backup withholding rules may be credited against a U.S. Holder’s U.S. tax liability, and a U.S. Holder may obtain a refund of any excess amounts withheld under the backup withholding rules by filing the appropriate claim for refund with the IRS.
115
Enforcement of Civil Liabilities
We are a public limited company incorporated under the laws of Australia. Certain of our directors are non-residents of the United States and substantially all of their assets are located outside the United States. As a result, it may not be possible or practicable for you to:
• effect service of process within the United States upon our non-U.S. resident directors or on us;
• enforce in U.S. courts judgments obtained against our non-U.S. resident directors or us in the United States courts in any action, including actions under the civil liability provisions of U.S. securities laws;
• enforce in U.S. courts judgments obtained against our non-U.S. resident directors or us in courts of jurisdictions outside the United States in any action, including actions under the civil liability provisions of U.S. securities laws; or
• bring an original action in an Australian court to enforce liabilities against our non-U.S. resident directors or us based solely upon U.S. securities laws.
You may also have difficulties enforcing in courts outside the United States judgments that are obtained in U.S. courts against any of our non-U.S. resident directors or us, including actions under the civil liability provisions of the U.S. securities laws.
With that noted, there are no treaties between Australia and the United States that would affect the recognition or enforcement of foreign judgments in Australia. We also note that investors may be able to bring an original action in an Australian court against us to enforce liabilities based in part upon U.S. federal securities laws. The disclosure in this section is not based on the opinion of counsel.
We have appointed Corporation Service Company as our agent to receive service of process with respect to any action brought against us under the federal securities laws of the United States.
116
We have entered into an underwriting agreement with the underwriter listed in the table below. We refer to the underwriter listed in the table below as the “underwriter.” Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to the underwriter, and the underwriter has agreed to purchase from us, ADSs of the Company. Prior to this offering, there has been no public markets for the ADSs. We have applied to list the ADSs on the Nasdaq Capital Market under the symbol “IXHL”.
Pursuant to the terms and subject to the conditions contained in the underwriting agreement, we have agreed to sell to the underwriter named below, and the underwriter has agreed to purchase from us, the number of ADSs set forth opposite its name below:
|
Underwriter |
Number of ADSs |
|
|
Roth Capital Partners, LLC |
||
|
Total |
The underwriting agreement provides that the obligation of the underwriter to purchase the ADSs offered by this prospectus is subject to certain conditions. The underwriter is obligated to purchase all of the ADSs offered hereby if any of the ADSs are purchased.
We have granted the underwriter an option to buy up to an additional ADSs from us at the public offering price, less the underwriting discounts and commissions, to cover over-allotments, if any. The underwriter may exercise this option at any time, in whole or in part, during the 30-day period after the date of this prospectus.
Discounts, Commissions and Expenses
The underwriter proposes to offer to the ADSs purchased pursuant to the underwriting agreement to the public at the public offering price set forth on the cover page of this prospectus and to certain dealers at that price less a concession not in excess of $ per ADS. After this offering, the public offering price and concession may be changed by the underwriter. No such change shall change the amount of proceeds to be received by us as set forth on the cover page of this prospectus.
In connection with the sale of the ADSs to be purchased by the underwriter, the underwriter will be deemed to have received compensation in the form of underwriting commissions and discounts. The underwriting commissions and discounts will be % of the gross proceeds of this offering, or $ per ADS, based on the public offering price per ADS set forth on the cover page of this prospectus.
We have also agreed to reimburse Roth Capital Partners at closing for expenses incurred by it in connection with the offering up to a maximum of $ .
The following table shows the underwriting discounts and commissions payable to the underwriter by us in connection with this offering (assuming both the exercise and non-exercise of the over-allotment option to purchase additional ADSs we have granted to the underwriter):
|
Per ADS |
Total |
|||||||||
|
Without |
With |
Without |
With |
|||||||
|
Public offering price |
$ |
$ |
||||||||
|
Underwriting discounts and commissions paid by us |
$ |
$ |
||||||||
Indemnification
Pursuant to the underwriting agreement, we have agreed to indemnify the underwriter against certain liabilities, including liabilities under the Securities Act, or to contribute to payments that the underwriter or such other indemnified parties may be required to make in respect of those liabilities.
117
Warrants
Upon the closing of this offering, we have agreed to sell to the underwriters a warrant to purchase up to 7.5% of the number of ordinary shares, represented by ADSs, sold in this offering. The warrant will be issued in three tranches, each comprising up to 2.5% of the number of shares of common stock sold in the offering, with the tranches exercisable at an exercise price equal to 120%, 135% and 150% of the public offering price per ADS sold pursuant to this offering, respectively, subject to standard anti-dilution adjustments for share splits and similar transactions. The warrant will be exercisable at any time, and from time to time, in whole or in part, during the period commencing 180 days from the commencement of sales in this offering, and expiring three years from the commencement of sales in this offering. The warrant is also exercisable on a cashless basis. The warrants have been deemed compensation by FINRA and are therefore subject to a 180-day lock-up pursuant to FINRA Rule 5110(e)(1). Except as permitted by Rule 5110(e)(1), the underwriter (or permitted assignees under the Rule) will not sell, transfer, assign, pledge, or hypothecate the warrants or the securities underlying the warrants, nor will any, of them engage in any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of the option or the underlying securities for a period of 180 days from the commencement of sales under this prospectus.
Lock-Up Agreements
We have agreed not to (i) offer, pledge, issue, sell, contract to sell, purchase, contract to purchase, lend or otherwise transfer or dispose of, directly or indirectly, any ADSs or any securities convertible into or exercisable or exchangeable for ADSs; (ii) enter into any swap or other arrangement that transfers, in whole or in part, any of the economic consequences of ownership of ADSs; or (iii) file any registration statement with the SEC relating to the offering of any ADSs or any securities convertible into or exercisable or exchangeable for ADSs, without the prior written consent of Roth Capital Partners for a period of 180 days following the date of this prospectus (the “Lock-up Period”). This consent may be given at any time without public notice. These restrictions on future issuances are subject to exceptions for (i) the issuance of ADSs sold in this offering, (ii) the issuance of ordinary shares or ADSs upon the exercise of options or warrants or the conversion of outstanding preferred stock or other outstanding convertible securities, or (iii) the issuance of employee stock options not exercisable during the Lock-Up Period.
In addition, each of our directors and executive officers has entered into a lock-up agreement with the underwriter. Under the lock-up agreements, the directors and executive officers may not, directly or indirectly, sell, offer to sell, contract to sell, or grant any option for the sale (including any short sale), grant any security interest in, pledge, hypothecate, hedge, establish an open “put equivalent position” (within the meaning of Rule 16a-1(h) under the Securities Exchange Act of 1934, as amended, or the Exchange Act), or otherwise dispose of, or enter into any transaction which is designed to or could be expected to result in the disposition of, any ADSs or securities convertible into or exchangeable for ADSs, or publicly announce any intention to do any of the foregoing, without the prior written consent of Roth Capital Partners, for a period of 180 days from the closing date of this offering. This consent may be given at any time without public notice. These restrictions on future dispositions by our directors and executive officers are subject to exceptions for (a) transfers (i) as a bona fide gift or gifts, provided that the donee or donees thereof agree to be bound in writing by the restrictions set forth in the lock-up agreement, or (ii) to any trust for the direct or indirect benefit of the undersigned or the immediate family of the undersigned, provided that the trustee of the trust agrees to be bound in writing by the restrictions of the lock-up agreement, and provided further that any such transfer shall not involve a disposition for value; or (b) the acquisition or exercise of any stock option approved by shareholders or issued pursuant to any equity incentive plan of the Company, limited only to options or plans that are described in this prospectus and provided the lock-up agreement applies to any of the securities issued upon such exercise.
Electronic Distribution
This prospectus may be made available in electronic format on websites or through other online services maintained by the underwriter or by its affiliates. In those cases, prospective investors may view offering terms online and prospective investors may be allowed to place orders online. Other than this prospectus in electronic format, the information on the underwriter’s website or our website and any information contained in any other websites maintained by the underwriter or by us is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or the underwriter in its capacity as underwriter, and should not be relied upon by investors.
118
Price Stabilization, Short Positions and Penalty Bids
In connection with the offering the underwriter may engage in stabilizing transactions, over-allotment transactions, syndicate covering transactions and penalty bids in accordance with Regulation M under the Exchange Act:
• Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum.
• Over-allotment involves sales by the underwriter of ADSs in excess of the number of ADSs the underwriter is obligated to purchase, which creates a syndicate short position. The short position may be either a covered short position or a naked short position. In a covered short position, the number of ADSs over-allotted by the underwriter is not greater than the number of ADSs that they may purchase in the over-allotment option. In a naked short position, the number of ADSs involved is greater than the number of ADSs in the over-allotment option. The underwriter may close out any covered short position by either exercising their over-allotment option and/or purchasing ADSs in the open market.
• Syndicate covering transactions involve purchases of the ADSs in the open market after the distribution has been completed in order to cover syndicate short positions. In determining the source of ADSs to close out the short position, the underwriter will consider, among other things, the price of ADSs available for purchase in the open market as compared to the price at which they may purchase ADSs through the over-allotment option. A naked short position occurs if the underwriter sells more ADSs than could be covered by the over-allotment option. This position can only be closed out by buying ADSs in the open market. A naked short position is more likely to be created if the underwriter is concerned that there could be downward pressure on the price of the ADSs in the open market after pricing that could adversely affect investors who purchase in the offering.
• Penalty bids permit the underwriter to reclaim a selling concession from a syndicate member when the ADSs originally sold by the syndicate member are purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions.
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of the ADSs or preventing or slowing a decline in the market price of the ADSs. As a result, the price of the ADSs may be higher than the price that might otherwise exist in the open market. These transactions may be discontinued at any time
Neither we nor the underwriter make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of the ADSs. In addition, neither we nor the underwriter make any representation that the underwriter will engage in these transactions or that any transaction, if commenced, will not be discontinued without notice.
Offer restrictions outside the United States
Other than in the United States, no action has been taken by us or the underwriter that would permit a public offering of the securities offered by this prospectus in any jurisdiction where action for that purpose is required. The securities offered by this prospectus may not be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sale of any such securities be distributed or published in any jurisdiction, except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons into whose possession this prospectus comes are advised to inform themselves about and to observe any restrictions relating to this offering and the distribution of this prospectus. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities offered by this prospectus in any jurisdiction in which such an offer or a solicitation is unlawful.
Notice to Prospective Investors in European Union
This prospectus has not been, and will not be, registered with or approved by any securities regulator in the European Union. Accordingly, this document may not be made available, nor may the ADSs be offered for sale, in the European Union except in circumstances that do not require a prospectus under Article 1(4) of Regulation (EU) 2017/1129 of the European Parliament and the Council of the European Union (the “Prospectus Regulation”).
119
In accordance with Article 1(4)(a) of the Prospectus Regulation, an offer of ADSs in the European Union is limited to persons who are “qualified investors” (as defined in Article 2(e) of the Prospectus Regulation).
Notice to Prospective Investors in the United Kingdom
Neither this prospectus nor any other document relating to the offer has been delivered for approval to the Financial Conduct Authority in the United Kingdom and no prospectus (within the meaning of section 85 of the Financial Services and Markets Act 2000, as amended (“FSMA”)) has been published or is intended to be published in respect of the ADSs.
The ADSs may not be offered or sold in the United Kingdom by means of this prospectus or any other document, except in circumstances that do not require the publication of a prospectus under section 86(1) of the FSMA. This prospectus is issued on a confidential basis in the United Kingdom to “qualified investors” within the meaning of Article 2(e) of the UK Prospectus Regulation. This prospectus may not be distributed or reproduced, in whole or in part, nor may its contents be disclosed by recipients, to any other person in the United Kingdom.
Each person in the UK who initially acquires any ADSs or to whom any offer is made will be deemed to have represented, acknowledged and agreed to and with the Company and the Underwriter that it is a qualified investor within the meaning of the UK Prospectus Regulation.
In the case of any ADSs being offered to a financial intermediary as that term is used in Article 5(1) of the UK Prospectus Regulation, each such financial intermediary will be deemed to have represented, acknowledged and agreed that the ADSs acquired by it in the offer have not been acquired on a non-discretionary basis on behalf of, nor have they been acquired with a view to their offer or resale to, persons in circumstances which may give rise to an offer to the public other than their offer or resale in the UK to qualified investors, in circumstances in which the prior consent of the representatives of the underwriters has been obtained to each such proposed offer or resale.
For the purposes of this provision, the expression “UK Prospectus Regulation” means Regulation (EU) 2017/1129 as it forms part of domestic law by virtue of the European Union (Withdrawal) Act 2018, and the expression “FSMA” means the Financial Services and Markets Act 2000.
In connection with the offering, the underwriters are not acting for anyone other than the issuer and will not be responsible to anyone other than the issuer for providing the protections afforded to their clients nor for providing advice in relation to the offering.
This document is for distribution only to persons who (i) have professional experience in matters relating to investments and who qualify as investment professionals within the meaning of Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005, or, as amended, the Financial Promotion Order, (ii) are persons falling within Article 49(2)(a) to (d), or high net worth companies, unincorporated associations etc., of the Financial Promotion Order, (iii) are outside the United Kingdom, or (iv) are persons to whom an invitation or inducement to engage in investment activity (within the meaning of Section 21 of the Financial Services and Markets Act 2000, as amended, or the FSMA) in connection with the issue or sale of any securities may otherwise lawfully be communicated or caused to be communicated, all such persons together being referred to as “relevant persons”. This document is directed only at relevant persons and must not be acted on or relied on by persons who are not relevant persons. Any investment or investment activity to which this document relates is available only to relevant persons and will be engaged in only with relevant persons.
Notice to Prospective Investors in Switzerland
The ADSs may not be publicly offered, directly or indirectly, in Switzerland within the meaning of the Swiss Financial Services Act (“FinSA”) and will not be listed or admitted to trading on the SIX Swiss Exchange or on any other trading venue (exchange or multilateral trading facility) in Switzerland. Neither this document nor any other offering or marketing material relating to the ADSs or the offering constitutes a prospectus as such term is understood pursuant to the FinSA. Neither this document nor any other offering or marketing material relating to the ADSs or the offering may be publicly distributed or otherwise made publicly available in Switzerland. The ADSs will be offered only to investors who qualify as “professional clients”, as defined in the FinSA.
120
Notice to Prospective Investors in Hong Kong
The ADSs have not been offered or sold and will not be offered or sold in Hong Kong, by means of any document, other than (a) to “professional investors” as defined in the Securities and Futures Ordinance (Cap. 571) of Hong Kong and any rules made under that Ordinance; or (b) in other circumstances which do not result in the document being a “prospectus” as defined in the Companies Ordinance (Cap. 32) of Hong Kong or which do not constitute an offer to the public within the meaning of that Ordinance. No advertisement, invitation or document relating to the ADSs has been or may be issued or has been or may be in the possession of any person for the purposes of issue, whether in Hong Kong or elsewhere, which is directed at, or the contents of which are likely to be accessed or read by, the public of Hong Kong (except if permitted to do so under the securities laws of Hong Kong) other than with respect to ADSs which are or are intended to be disposed of only to persons outside Hong Kong or only to “professional investors” as defined in the Securities and Futures Ordinance and any rules made under that Ordinance.
Notice to Prospective Investors in Japan
The ADSs have not been and will not be registered under the Financial Instruments and Exchange Law of Japan (Law No. 25 of 1948, as amended (“FIEL”) and, accordingly, will not be offered or sold, directly or indirectly, in Japan, or for the benefit of any Japanese person or to others for re-offering or resale, directly or indirectly, in Japan or to any Japanese person, except to Qualified Institutional Investors as defined in the FIEL in compliance with all applicable laws, regulations and ministerial guidelines promulgated by relevant Japanese governmental or regulatory authorities in effect at the relevant time.
Notice to Prospective Investors in Singapore
This prospectus has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, the ADSs were not offered or sold or caused to be made the subject of an invitation for subscription or purchase and will not be offered or sold or caused to be made the subject of an invitation for subscription or purchase, and this prospectus or any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of the ADSs, has not been circulated or distributed, nor will it be circulated or distributed, whether directly or indirectly, to any person in Singapore other than (i) to an institutional investor (as defined in Section 4A of the Securities and Futures Act (Chapter 289) of Singapore, as modified or amended from time to time (the “SFA”)) pursuant to Section 274 of the SFA or (ii) to an “accredited investor” (as defined in Section 4A of the SFA) pursuant to Section 275(1) of the SFA.
121
Expenses Relating to the Offering
The following table sets forth the costs and expenses, other than underwriting discounts and commissions, payable in connection with the sale of ADSs in the offering. All amounts are estimated except the SEC registration fee, the Financial Industry Regulatory Authority, Inc., or FINRA, filing fee and the Nasdaq initial listing fee. Except as otherwise noted, all the expenses below will be paid by us.
|
Expense |
Amount |
|
|
SEC registration fee |
US$2,935 |
|
|
FINRA filing fee |
* |
|
|
Nasdaq initial listing fee |
* |
|
|
Legal fees and expenses |
* |
|
|
Accounting fees and expenses |
* |
|
|
Printing expenses |
* |
|
|
Miscellaneous fees and expenses |
* |
|
|
Total |
US$* |
____________
* To be provided by amendment.
122
The validity of the ordinary shares represented by the ADSs and certain other matters of Australian law will be passed upon for us by Rimôn Law Pty Ltd. Certain matters as to U.S. federal law and New York state law will be passed upon for us by Rimôn Law Pty Ltd. Legal counsel to the underwriter in connection with this offering are Faegre Drinker Biddle & Reath LLP, with respect to U.S. federal law.
The consolidated financial statements of Incannex Healthcare Limited as of June 30, 2020, and 2019, and for the years appearing in the prospectus have been audited by WithumSmith+Brown, PC (“Withum”), independent registered public accounting firm, as set forth in their report thereon which includes an explanatory paragraph relating to the Incannex Healthcare Limited’s ability to continue as a going concern, relating to the consolidated financial statements of the Company, appearing elsewhere in the prospectus, and are included in reliance on the report of such firm given upon their authority as experts in accounting and auditing.
The offices of Withum are located at 1411 Broadway 9th floor, New York, NY 10018.
Where You Can Find More Information
We have filed with the SEC a Registration Statement on Form F-1 under the Securities Act with respect to the ADSs offered in this prospectus. A related registration statement on Form F-6 has been filed with the SEC to register the ADSs. This prospectus, which forms a part of the Registration Statement, does not contain all of the information included in the Registration Statement. Certain information is omitted and you should refer to the Registration Statement and its exhibits for that information. With respect to references made in this prospectus to any contract or other document of Incannex, such references are not necessarily complete and you should refer to the exhibits attached to the Registration Statement for copies of the actual contract or document.
Upon the closing of this offering, we will become subject to periodic reporting and other informational requirements of the Exchange Act as applicable to foreign private issuers. Accordingly, we will be required to file reports, including annual reports on Form 20-F, periodic reports and other information, with the SEC.
We are allowed four months after the end of our fiscal year to file our annual report with the SEC, and we are not required to disclose certain detailed information regarding executive compensation that is required from U.S. domestic issuers. Also, as a foreign private issuer, we are exempt from the rules of the Exchange Act prescribing the furnishing of proxy statements to shareholders, and the members of our board of directors, our senior management and our principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act.
The SEC maintains a website at www.sec.gov that contains reports, proxy and information statements and other information regarding registrants that file electronically with the SEC. You also can inspect our registration statement, as well as any other information we file with or furnish to the SEC, on this website. This reference to the SEC’s website is an inactive textual reference only and is not a hyperlink.
We expect to make our annual reports and other information filed with or furnished to the SEC available, free of charge, through our website at www.incannex.com.au as soon as reasonably practicable after those reports and other information are filed with or furnished to the SEC. The information contained on, or that can be accessed through, our website is not part of, and is not incorporated into, this prospectus.
123
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
|
Page |
||
|
Consolidated Financial Statements for the years ended June 30, 2020 and 2019 |
||
|
F-2 |
||
|
F-3 |
||
|
F-4 |
||
|
F-5 |
||
|
F-6 |
||
|
F-7 |
INDEX TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
|
F-36 |
||
|
F-37 |
||
|
F-38 |
||
|
F-39 |
||
|
F-40 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors of
Incannex Healthcare Limited:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated statements of financial position of Incannex Healthcare Limited (the “Company”) as of 30 June 2020 and 2019, the related consolidated statements of comprehensive income/(loss), changes in equity and cash flows, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the consolidated financial position of the Company as of 30 June 2020 and 2019, and the consolidated results of its operations and its cash flows for the years then ended, in conformity with International Financial Reporting Standards (IFRS) as issued by the International Accounting Standards Board (IASB).
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has negative cash flows from operations, continuing losses, and has been impacted by the COVID-19 crisis. As such there is substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ WithumSmith+Brown, PC
We have served as the Company’s auditor since 2021.
New York, New York
August 17, 2021
F-2
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME/(LOSS)
For the years ended 30 June 2020 and 2019
|
Notes |
2020 |
2019 |
||||||
|
$ |
$ |
|||||||
|
Revenue |
3 |
604,884 |
|
— |
|
|||
|
Other income |
3 |
217,170 |
|
1,553 |
|
|||
|
Product costs |
1 |
(450,345 |
) |
— |
|
|||
|
Administration expense |
1 |
(457,673 |
) |
(330,178 |
) |
|||
|
Advertising and promotion |
1 |
(406,225 |
) |
(94,814 |
) |
|||
|
Research and development costs |
1 |
(2,110,639 |
) |
(736,140 |
) |
|||
|
Compliance, legal and regulatory |
1 |
(235,163 |
) |
(72,181 |
) |
|||
|
Finance cost |
— |
|
(85,065 |
) |
||||
|
Share based payments |
13 |
(565,448 |
) |
(47,854 |
) |
|||
|
Occupancy expenses |
1 |
(2,085 |
) |
(1,519 |
) |
|||
|
Salaries and employee benefit expense |
1 |
(523,760 |
) |
(60,000 |
) |
|||
|
|
|
|||||||
|
Loss before tax from continuing operations |
(3,929,284 |
) |
(1,426,198 |
) |
||||
|
Income tax benefit |
5 |
— |
|
— |
|
|||
|
Loss after tax from continuing operations |
(3,929,284 |
) |
(1,426,198 |
) |
||||
|
Loss on discontinued operations, net of tax |
6 |
(768,352 |
) |
(1,292,201 |
) |
|||
|
|
|
|||||||
|
Total comprehensive loss |
(4,697,636 |
) |
(2,718,399 |
) |
||||
|
|
|
|||||||
|
Basic loss per share from continuing and discontinued operations (cents per share) |
7 |
(0.69 |
) |
(0.40 |
) |
|||
|
|
|
|||||||
|
Basic loss per share from continuing operations (cents per share) |
7 |
(0.57 |
) |
(0.21 |
) |
|||
The accompanying notes form part of these financial statements
F-3
CONSOLIDATED STATEMENTS OF FINANCIAL POSITION
As at 30 June 2020 and 2019
|
Notes |
2020 |
2019 |
||||||
|
$ |
$ |
|||||||
|
Assets |
|
|
||||||
|
Current assets |
|
|
||||||
|
Cash |
9 |
3,603,390 |
|
93,332 |
|
|||
|
Trade and other receivables |
10 |
413,268 |
|
97,784 |
|
|||
|
Other assets |
12 |
36,262 |
|
39,191 |
|
|||
|
Inventory |
14 |
183,159 |
|
152,804 |
|
|||
|
Total current assets |
4,236,079 |
|
383,111 |
|
||||
|
Non-current assets |
|
|
||||||
|
Intangible assets |
15 |
— |
|
49,377 |
|
|||
|
Property, plant and equipment |
11 |
— |
|
85,423 |
|
|||
|
Total non-current assets |
— |
|
134,800 |
|
||||
|
Total assets |
4,236,079 |
|
517,911 |
|
||||
|
Liabilities |
|
|
||||||
|
Current liabilities |
|
|
||||||
|
Trade and other payables |
16 |
955,006 |
|
478,820 |
|
|||
|
Borrowings from related party |
— |
|
65,000 |
|
||||
|
Other liabilities |
17 |
116,645 |
|
391,271 |
|
|||
|
Total current liabilities |
1,071,651 |
|
935,091 |
|
||||
|
|
|
|||||||
|
Total liabilities |
1,071,651 |
|
935,091 |
|
||||
|
Net assets/(liabilities) |
3,164,428 |
|
(417,180 |
) |
||||
|
Equity attributable to owners of the parent |
|
|
||||||
|
Issued capital |
18 |
34,192,043 |
|
26,951,744 |
|
|||
|
Reserves |
19 |
1,490,588 |
|
451,643 |
|
|||
|
Accumulated losses |
(32,518,203 |
) |
(27,820,567 |
) |
||||
|
Net equity |
3,164,428 |
|
(417,180 |
) |
||||
The accompanying notes form part of these financial statements
F-4
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
For the years ended 30 June 2020 and 2019
|
Consolidated |
Issued |
Equity |
Accumulated Losses |
Total |
|||||||
|
$ |
$ |
$ |
$ |
||||||||
|
Balance at 1 July 2018 |
24,410,905 |
|
229,725 |
(25,022,948 |
) |
(382,318 |
) |
||||
|
Adjustment on initial application of IFRS15 |
— |
|
— |
(79,220 |
) |
(79,220 |
) |
||||
|
Comprehensive loss for the year |
— |
|
— |
(2,718,399 |
) |
(2,718,399 |
) |
||||
|
Options issued to advisors |
— |
|
221,918 |
— |
|
221,918 |
|
||||
|
Shares issued |
2,914,248 |
|
— |
— |
|
2,914,248 |
|
||||
|
Shares issue costs |
(373,409 |
) |
— |
— |
|
(373,409 |
) |
||||
|
Balance at 30 June 2019 |
26,951,744 |
|
451,643 |
(27,820,567 |
) |
(417,180 |
) |
||||
|
|
|
|
|||||||||
|
Balance at 30 June 2019 |
26,951,744 |
|
451,643 |
(27,820,567 |
) |
(417,180 |
) |
||||
|
Comprehensive loss for the year |
— |
|
— |
(4,697,636 |
) |
(4,697,636 |
) |
||||
|
Options exercised |
1,077,093 |
|
— |
— |
|
1,077,093 |
|
||||
|
Options issued to advisors |
— |
|
449,093 |
— |
|
449,093 |
|
||||
|
Share based payments |
— |
|
589,852 |
— |
|
589,852 |
|
||||
|
Shares issued |
7,105,354 |
|
— |
— |
|
7,105,354 |
|
||||
|
Shares issue costs |
(942,148 |
) |
— |
— |
|
(942,148 |
) |
||||
|
Balance at 30 June 2020 |
34,192,043 |
|
1,490,588 |
(32,518,203 |
) |
3,164,428 |
|
||||
The accompanying notes form part of these financial statements
F-5
CONSOLIDATED STATEMENTS OF CASH FLOWS
For the years ended 30 June 2020 and 2019
|
Notes |
2020 |
2019 |
||||||
|
$ |
$ |
|||||||
|
Cash flows from operating activities |
|
|
||||||
|
Receipts from customers |
1,172,084 |
|
1,148,410 |
|
||||
|
Receipts from other income |
217,170 |
|
1,553 |
|
||||
|
Payments to suppliers and employees |
(5,299,667 |
) |
(3,371,103 |
) |
||||
|
Interest received |
3,079 |
|
1,633 |
|
||||
|
Finance costs paid |
— |
|
(92,249 |
) |
||||
|
Research and development tax refund |
— |
|
151,323 |
|
||||
|
Net cash used in operating activities |
9(i) |
(3,907,334 |
) |
(2,160,433 |
) |
|||
|
|
|
|||||||
|
Cash flows from investing activities |
|
|
||||||
|
Payments for property, plant and equipment |
— |
|
(24,442 |
) |
||||
|
Proceeds from disposal of property, plant and equipment |
13,000 |
|
— |
|
||||
|
Net cash provided by/(used in) investing activities |
13,000 |
|
(22,942 |
) |
||||
|
|
|
|||||||
|
Cash flows from financing activities |
|
|
||||||
|
Proceeds from shares issued (net of costs) |
7,469,392 |
|
2,184,801 |
|
||||
|
Debt repaid |
(65,000 |
) |
(200,000 |
) |
||||
|
Proceeds from borrowing |
— |
|
65,000 |
|
||||
|
Net cash provided by financing activities |
7,404,392 |
|
2,049,801 |
|
||||
|
|
|
|||||||
|
Net increase/(decrease) in cash |
3,510,058 |
|
(135,074 |
) |
||||
|
Cash at beginning of the year |
93,332 |
|
228,406 |
|
||||
|
Cash at end of the year |
9 |
3,603,390 |
|
93,332 |
|
|||
The consolidated statement of cash flows above presents the total cash flows of the Company, inclusive of discontinued operations. The cash flows from discontinued operations for the years ended 30 June 2020 and 30 June 2019 are as follows:
• Cash flows used in operating activities: ($636,857) in 2020 and ($1,154,399) in 2019;
• Cash flows from investing activities: $13,000 in 2020 and nil in 2019;
• Cash flows used in financing activities: nil in 2020 and nil in 2019;
The accompanying notes form part of these financial statements
F-6
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies
The principal accounting policies adopted in the preparation of the consolidated financial statements are set out below. These policies have been consistently applied to all the years presented, unless otherwise stated.
Nature of Operations and Going Concern
Incannex Healthcare Limited (the “Company”) and its consolidated subsidiaries (collectively, the “Group”) is a clinical stage pharmaceutical development company that is developing unique medicinal cannabis pharmaceutical products and psychedelic medicine therapies. The Company’s common shares trade on the Australian Stock Market (“ASX”). The Company’s resgistered office is at Suite 15, Level 12, 401 Docklands Drive, Docklands 3008, Victoria, Australia.
For the six months ended 31 December 2020, the Group incurred a total comprehensive loss after income tax of $3.1 million and had net cash outflows from operations of $2.8 million. The Group held total cash of $11.8 million as of 31 December 2020.
Capital raising will be required for us to meet our forecast expenditure and continue as a going concern, although there is uncertainty related to these cash inflows because the ability to access external funding is not wholly within the Group’s control.
Management and the directors believe that the Group will be successful in the above matters and, accordingly, have prepared the financial report on a going concern basis, notwithstanding that if losses continue, and we are unable raise additional financing on sufficiently attractive terms, then we may not have sufficient liquidity to sustain our operations and may not be able to continue as a going concern.
New or amended Accounting Standards and Interpretations adopted
The consolidated entity has adopted all of the new or amended Accounting Standards and Interpretations issued by the International Accounting Standards Board (‘IASB’) that are mandatory for the current reporting periods.
Any new or amended Accounting Standards or Interpretations that are not yet mandatory have not been early adopted.
Historical cost convention
The financial statements have been prepared under the historical cost convention, except for, where applicable, the revaluation of financial assets and liabilities at fair value through profit or loss, financial assets at fair value through other comprehensive income, investment properties, certain classes of property, plant and equipment and derivative financial instruments.
Critical accounting estimates
The preparation of the financial statements requires the use of certain critical accounting estimates. It also requires management to exercise its judgement in the process of applying the consolidated entity’s accounting policies. The areas involving a higher degree of judgement or complexity, or areas where assumptions and estimates are significant to the financial statements, are disclosed in note 2.
Statement of compliance
These financial statements were authorised for issue by the Board of Directors on 17 August 2021.
The financial statements comply with International Financial Reporting Standards (IFRS), as issued by the International Accounting Standards Board (IASB).
F-7
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
Parent entity information
In accordance with IFRS 10 Consolidated Financial Statements, these financial statements present the results of the consolidated entity only. Supplementary information about the parent entity is disclosed in note 26.
Principles of consolidation
The consolidated financial statements incorporate the assets and liabilities of all subsidiaries of Incannex Healthcare Limited (‘Company’ or ‘parent entity’) as at 30 June 2019 and 2020 and the results of all subsidiaries for the years then ended. Incannex Healthcare Limited and its subsidiaries together are referred to in these financial statements as the ‘consolidated entity’. Details of all controlled entities are set out in Note 24.
Subsidiaries are all those entities over which the consolidated entity has control. The consolidated entity controls an entity when the consolidated entity is exposed to, or has rights to, variable returns from its involvement with the entity and has the ability to affect those returns through its power to direct the activities of the entity. Subsidiaries are fully consolidated from the date on which control is transferred to the consolidated entity. They are de-consolidated from the date that control ceases.
Intercompany transactions, balances and unrealised gains on transactions between entities in the consolidated entity are eliminated. Unrealised losses are also eliminated unless the transaction provides evidence of the impairment of the asset transferred. Accounting policies of subsidiaries have been changed where necessary to ensure consistency with the policies adopted by the consolidated entity.
Where the consolidated entity loses control over a subsidiary, it derecognises the assets including goodwill, liabilities and non-controlling interest in the subsidiary together with any cumulative translation differences recognised in equity. The consolidated entity recognises the fair value of the consideration received and the fair value of any investment retained together with any gain or loss in profit or loss.
Operating segments
Operating segments are presented using the ‘management approach’, where the information presented is on the same basis as the internal reports provided to the Chief Executive Officer. The Chief Executive Officer is responsible for the allocation of resources to operating segments and assessing their performance.
Foreign currency translation
The financial statements are presented in Australian dollars, which is Incannex Healthcare Limited’s functional and presentation currency.
Foreign currency transactions
Foreign currency transactions are translated into Australian dollars using the exchange rates prevailing at the dates of the transactions. Foreign exchange gains and losses resulting from the settlement of such transactions and from the translation at financial year-end exchange rates of monetary assets and liabilities denominated in foreign currencies are recognised in profit or loss.
Revenue recognition
The Company’s revenues were generated from the sale of pharmaceutical Medicinal Cannabis products through the Special Access Scheme in Australia. Revenue comprises the fair value of the consideration received, or receivable and it is shown net of tax and discounts. The Company also earned revenue from the sale of dentist products through e-commerce website, however, the Company discontinued this segment on 30 June 2020.
F-8
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
The Company recognizes revenue to depict the transfer of goods and services to clients in an amount that reflects the consideration to which the Company expects to be entitled in exchange for those goods and services by applying the following steps:
• Identify the contract with a client;
• Identify the performance obligations in the contract;
• Determine the transaction price;
• Allocate the transaction price to the performance obligations; and
• Recognize revenue when, or as, the Company satisfies a performance obligation.
Revenue may be earned over time as the performance obligations are satisfied or at a point in time which is when the entity has earned a right to payment, the customer has possession of the asset and the related significant risks and rewards of ownership, and the customer has accepted the asset.
The Company’s arrangements with clients can include multiple performance obligations. When contracts involve various performance obligations, the Company evaluates whether each performance obligation is distinct and should be accounted for as a separate unit of accounting under IFRS 15, Revenue from Contracts with Customers.
The Company determines the standalone selling price by considering its overall pricing objectives and market conditions. Significant pricing practices taken into consideration include discounting practices, the size and volume of our transactions, our marketing strategy, historical sales, and contract prices. The determination of standalone selling prices is made through consultation with and approval by management, taking into consideration our go-to-market strategy. As the Company’s go-to-market strategies evolve, the Company may modify its pricing practices in the future, which could result in changes in relative standalone selling prices.
The Company disaggregates revenue from contracts with customers based on the categories that most closely depict how the nature, amount, timing and uncertainty of revenue and cash flows are affected by economic factors. During the years ended 30 June 2020 and 2019, the Company recognized revenue from only one such category, being cannabinoid oils sales. As stated in Note 4 to these financial statements, the Company previously recognized revenue from oral and dental devices, although these operations have been discontinued. All sales are made within Australia and the Company has not disaggregated revenue based on geography.
The Company receives payment from its clients after invoicing within the normal 28-day commercial terms. If a client is specifically identified as a credit risk, recognition of revenue is stopped except to the extent of fees that have already been collected.
Interest and Other income
Interest revenue is recognised when it is received or when the right to receive it is established.
Income tax
The income tax expense or benefit for the period is the tax payable on that period’s taxable income based on the applicable income tax rate for each jurisdiction, adjusted by the changes in deferred tax assets and liabilities attributable to temporary differences, unused tax losses and the adjustment recognised for prior periods, where applicable.
Deferred tax assets and liabilities are recognised for temporary differences at the tax rates expected to be applied when the assets are recovered or liabilities are settled, based on those tax rates that are enacted or substantively enacted, except for:
F-9
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
• When the deferred income tax asset or liability arises from the initial recognition of goodwill or an asset or liability in a transaction that is not a business combination and that, at the time of the transaction, affects neither the accounting nor taxable profits; or
• When the taxable temporary difference is associated with interests in subsidiaries, associates or joint ventures, and the timing of the reversal can be controlled, and it is probable that the temporary difference will not reverse in the foreseeable future.
Deferred tax assets are recognised for deductible temporary differences and unused tax losses only if it is probable that future taxable amounts will be available to utilise those temporary differences and losses.
The carrying amount of recognised and unrecognised deferred tax assets are reviewed at each reporting date. Deferred tax assets recognised are reduced to the extent that it is no longer probable that future taxable profits will be available for the carrying amount to be recovered. Previously unrecognised deferred tax assets are recognised to the extent that it is probable that there are future taxable profits available to recover the asset.
Deferred tax assets and liabilities are offset only where there is a legally enforceable right to offset current tax assets against current tax liabilities and deferred tax assets against deferred tax liabilities; and they relate to the same taxable authority on either the same taxable entity or different taxable entities which intend to settle simultaneously.
Discontinued operations
A discontinued operation is a component of the consolidated entity that has been disposed of or is classified as held for sale and that represents a separate major line of business or geographical area of operations, is part of a single co-ordinated plan to dispose of such a line of business or area of operations, or is a subsidiary acquired exclusively with a view to resale. The results of discontinued operations are presented separately on the face of the statement of comprehensive income.
Government grants
Income from government grants is recognised only when the Company has reasonable assurance that the grants will be received, and the conditions of the grants will be complied with. Income from Government grants is recognised on a systematic basis using the income approach over the periods in which the Company recognises as expenses the related costs for which the grants are intended to compensate.
Current and non-current classification
Assets and liabilities are presented in the statement of financial position based on current and non-current classification.
An asset is classified as current when: it is either expected to be realised or intended to be sold or consumed in the consolidated entity’s normal operating cycle; it is held primarily for the purpose of trading; it is expected to be realised within 12 months after the reporting period; or the asset is cash or cash equivalent unless restricted from being exchanged or used to settle a liability for at least 12 months after the reporting period. All other assets are classified as non-current.
A liability is classified as current when: it is either expected to be settled in the consolidated entity’s normal operating cycle; it is held primarily for the purpose of trading; it is due to be settled within 12 months after the reporting period; or there is no unconditional right to defer the settlement of the liability for at least 12 months after the reporting period. All other liabilities are classified as non-current.
Deferred tax assets and liabilities are classified as non-current.
F-10
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
Cash
Cash and deposits held at call with financial institutions, other short-term, highly liquid investments with original maturities of three months or less that are readily convertible to known amounts of cash and which are subject to an insignificant risk of changes in value. For the statement of cash flows presentation purposes, cash also includes bank overdrafts, which are shown within borrowings in current liabilities on the statement of financial position.
Trade and other receivables
Trade receivables are initially recognised at fair value and subsequently measured at amortised cost using the effective interest method, less any allowance for expected credit losses. Trade receivables are due for settlement within 30 days.
The consolidated entity has applied the simplified approach to measuring expected credit losses, which uses a lifetime expected loss allowance. To measure the expected credit losses, trade receivables have been grouped based on days overdue.
Other receivables are recognised at amortised cost, less any allowance for expected credit losses.
Inventory
Inventory Raw materials, work in progress and finished goods are stated at the lower of cost and net realisable value on a ‘first in first out’ basis. Cost comprises of direct materials and delivery costs, direct labour, import duties and other taxes, an appropriate proportion of variable and fixed overhead expenditure based on normal operating capacity, and, where applicable, transfers from cash flow hedging reserves in equity. Costs of purchased inventory are determined after deducting rebates and discounts received or receivable.
Stock in transit is stated at the lower of cost and net realisable value. Cost comprises of purchase and delivery costs, net of rebates and discounts received or receivable.
Net realisable value is the estimated selling price in the ordinary course of business less the estimated costs of completion and the estimated costs necessary to make the sale.
Other financial assets
Other financial assets are initially measured at fair value. Transaction costs are included as part of the initial measurement, except for financial assets at fair value through profit or loss. Such assets are subsequently measured at either amortised cost or fair value depending on their classification. Classification is determined based on both the business model within which such assets are held and the contractual cash flow characteristics of the financial asset unless an accounting mismatch is being avoided.
Financial assets are derecognised when the rights to receive cash flows have expired or have been transferred and the consolidated entity has transferred substantially all the risks and rewards of ownership. When there is no reasonable expectation of recovering part or all a financial asset, its carrying value is written off.
Financial assets at fair value through profit or loss
Financial assets not measured at amortised cost or at fair value through other comprehensive income are classified as financial assets at fair value through profit or loss. Typically, such financial assets will be either: (i) held for trading, where they are acquired for the purpose of selling in the short-term with an intention of making a profit, or a derivative; or (ii) designated as such upon initial recognition where permitted. Fair value movements are recognised in profit or loss.
F-11
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
Financial assets at fair value through other comprehensive income
Financial assets at fair value through other comprehensive income include equity investments which the consolidated entity intends to hold for the foreseeable future and has irrevocably elected to classify them as such upon initial recognition.
Impairment of financial assets
The consolidated entity recognises a loss allowance for expected credit losses on financial assets which are either measured at amortised cost or fair value through other comprehensive income. The measurement of the loss allowance depends upon the consolidated entity’s assessment at the end of each reporting period as to whether the financial instrument’s credit risk has increased significantly since initial recognition, based on reasonable and supportable information that is available, without undue cost or effort to obtain.
Where there has not been a significant increase in exposure to credit risk since initial recognition, a 12-month expected credit loss allowance is estimated. This represents a portion of the asset’s lifetime expected credit losses that is attributable to a default event that is possible within the next 12 months. Where a financial asset has become credit impaired or where it is determined that credit risk has increased significantly, the loss allowance is based on the asset’s lifetime expected credit losses. The amount of expected credit loss recognised is measured on the basis of the probability weighted present value of anticipated cash shortfalls over the life of the instrument discounted at the original effective interest rate.
For financial assets mandatorily measured at fair value through other comprehensive income, the loss allowance is recognised in other comprehensive income with a corresponding expense through profit or loss. In all other cases, the loss allowance reduces the asset’s carrying value with a corresponding expense through profit or loss.
Impairment of non-financial assets
Non-financial assets are subject to impairment test whenever events or changes in circumstances indicate that their carrying amount may not be recoverable. Where the carrying value of the non-financial asset exceeds its recoverable amount (i.e. the higher of value in use and fair value less costs to dispose), the asset is written down and impairment charge is recognized accordingly.
Where it is not possible to estimate the recoverable amount of an individual asset, the impairment test is carried out on the asset’s cash-generating unit (i.e. the smallest group of assets to which the asset belongs that generates cash inflow that is largely independent of cash inflows from other assets).
An impairment loss allocated to an asset, is reversed only if there have been changes in the estimates used to determine the asset’s recoverable amount since the last impairment loss was recognized.
Reversal of an impairment loss, as above, is limited to the lower of the carrying amount of the asset that would have been determined (net of depreciation or amortization) had no impairment loss been recognized for the asset in prior years and the asset’s recoverable amount. After an impairment of non-financial asset is recognized, the Company examines at each reporting date whether there are indications that the impairment which was recognized in the past no longer exists or should be reduced. The reversal of impairment loss of an asset is recognized in profit or loss.
Property, plant and equipment
Plant and equipment are stated at historical cost less accumulated depreciation and impairment. Historical cost includes expenditure that is directly attributable to the acquisition of the items.
F-12
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
Depreciation is calculated on a straight-line basis to write off the net cost of each item of property, plant, and equipment (excluding land) over their expected useful lives as follows:
|
Buildings |
40 years |
|||
|
Leasehold improvements |
3–10 years |
|||
|
Plant and equipment |
3–7 years |
The residual values, useful lives and depreciation methods are reviewed, and adjusted if appropriate, at each reporting date.
Leasehold improvements are depreciated over the unexpired period of the lease or the estimated useful life of the assets, whichever is shorter.
An item of property, plant and equipment is derecognised upon disposal or when there is no future economic benefit to the consolidated entity. Gains and losses between the carrying amount and the disposal proceeds are taken to profit or loss. Any revaluation surplus reserve relating to the item disposed of is transferred directly to equity.
Intangible assets
Research and development
Research costs are expensed in the period in which they are incurred. Development costs are capitalised when it is probable that the project will be a success considering its commercial and technical feasibility; the consolidated entity is able to use or sell the asset; the consolidated entity has sufficient resources and intent to complete the development; and its costs can be measured reliably. Capitalised development costs are amortised on a straight-line basis over the period of their expected benefit, being their finite life of 10 years.
Patents and trademarks
Significant costs associated with patents and trademarks are deferred and amortised on a straight-line basis over the period of their expected benefit, being their finite life of 10 years.
Trade and other payables
These amounts represent liabilities for goods and services provided to the consolidated entity prior to the end of the financial years and which are unpaid. Due to their short-term nature, they are measured at amortised cost and are not discounted. The amounts are unsecured and are usually paid within 30 days of recognition.
Borrowings
Loans and borrowings are initially recognised at the fair value of the consideration received, net of transaction costs. They are subsequently measured at amortised cost using the effective interest method.
Lease liabilities
A lease liability is recognised at the commencement date of a lease. The lease liability is initially recognised at the present value of the lease payments to be made over the term of the lease, discounted using the interest rate implicit in the lease or, if that rate cannot be readily determined, the consolidated entity’s incremental borrowing rate. Lease payments comprise of fixed payments less any lease incentives receivable, variable lease payments that depend on an index or a rate, amounts expected to be paid under residual value guarantees, exercise price of a purchase option when the exercise of the option is reasonably certain to occur, and any anticipated termination penalties. The variable lease payments that do not depend on an index or a rate are expensed in the period in which they are incurred.
F-13
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
Lease liabilities are measured at amortised cost using the effective interest method. The carrying amounts are remeasured if there is a change in the following: future lease payments arising from a change in an index, or a rate used; residual guarantee; lease term; certainty of a purchase option and termination penalties. When a lease liability is remeasured, an adjustment is made to the corresponding right-of use asset, or to profit or loss if the carrying amount of the right-of-use asset is fully written down.
No lease liabilities are recognized for leases where the lease term is 12 months or less at the commencement date and for leases where the underlying value is deemed to be of low value. The costs of any such leases are recorded within expenses as incurred.
Finance costs
Finance costs attributable to qualifying assets are capitalised as part of the asset. All other finance costs are expensed in the period in which they are incurred.
Provisions
Provisions are recognised when the consolidated entity has a present (legal or constructive) obligation as a result of a past event, it is probable the consolidated entity will be required to settle the obligation, and a reliable estimate can be made of the amount of the obligation. The amount recognised as a provision is the best estimate of the consideration required to settle the present obligation at the reporting date, taking into account the risks and uncertainties surrounding the obligation. If the time value of money is material, provisions are discounted using a current pre-tax rate specific to the liability. The increase in the provision resulting from the passage of time is recognised as a finance cost.
Employee benefits
Short-term employee benefits
Liabilities for wages and salaries, including non-monetary benefits, annual leave and long service leave expected to be settled wholly within 12 months of the reporting date are measured at the amounts expected to be paid when the liabilities are settled.
Other long-term employee benefits
The liability for annual leave and long service leave not expected to be settled within 12 months of the reporting date are measured at the present value of expected future payments to be made in respect of services provided by employees up to the reporting date using the projected unit credit method. Consideration is given to expected future wage and salary levels, experience of employee departures and periods of service. Expected future payments are discounted using market yields at the reporting date on corporate bonds with terms to maturity and currency that match, as closely as possible, the estimated future cash outflows.
Share-based payments
Equity-settled compensation benefits are provided to employees.
Equity-settled transactions are awards of shares, performance rights or options over shares, that are provided to employees in exchange for the rendering of services.
The cost of equity-settled transactions are measured at fair value on grant date. Fair value is independently determined using either the trinomial or Black-Scholes option pricing model that takes into account the exercise price, the term of the option, the impact of dilution, the share price at grant date and expected price volatility of the underlying share, the expected dividend yield and the risk free interest rate for the term of the option, together with non-vesting conditions that do not determine whether the consolidated entity receives the services that entitle the
F-14
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
employees to receive payment. Inputs into the trinomial and Black-Scholes option pricing models used to calculate fair value are classified as level three inputs under the fair value hierarchy of IFRS 13. No account is taken of any other vesting conditions.
The cost of equity-settled transactions are recognised as an expense with a corresponding increase in equity over the vesting period. The cumulative charge to profit or loss is calculated based on the grant date fair value of the award, the best estimate of the number of awards that are likely to vest and the expired portion of the vesting period. The amount recognised in profit or loss for the period is the cumulative amount calculated at each reporting date less amounts already recognised in previous periods.
Market conditions are taken into consideration in determining fair value. Therefore any awards subject to market conditions are considered to vest irrespective of whether or not that market condition has been met, provided all other conditions are satisfied.
If equity-settled awards are modified, as a minimum an expense is recognised as if the modification has not been made. An additional expense is recognised, over the remaining vesting period, for any modification that increases the total fair value of the share-based compensation benefit as at the date of modification.
If the non-vesting condition is within the control of the consolidated entity or employee, the failure to satisfy the condition is treated as a cancellation. If the condition is not within the control of the consolidated entity or employee and is not satisfied during the vesting period, any remaining expense for the award is recognised over the remaining vesting period, unless the award is forfeited.
If equity-settled awards are cancelled, it is treated as if it has vested on the date of cancellation, and any remaining expense is recognised immediately. If a new replacement award is substituted for the cancelled award, the cancelled and new award is treated as if they were a modification.
Fair value measurement
When an asset, liability or equity instrument, financial or non-financial, is measured at fair value for recognition or disclosure purposes, the fair value is based on the price that would be received to sell an asset or an equity instrument or paid to transfer a liability in an orderly transaction between market participants at the measurement date; and assumes that the transaction will take place either: in the principal market; or in the absence of a principal market, in the most advantageous market.
Fair value is measured using the assumptions that market participants would use when pricing the asset, liability or equity instrument, assuming they act in their economic best interests. For non-financial assets, the fair value measurement is based on its highest and best use. Valuation techniques that are appropriate in the circumstances and for which sufficient data are available to measure fair value, are used, maximising the use of relevant observable inputs and minimising the use of unobservable inputs.
Assets, liabilities and equity instruments measured at fair value are classified into three levels, using a fair value hierarchy that reflects the significance of the inputs used in making the measurements. For assets and liabilities measured at fair value after initial recognition, classifications are reviewed at each reporting date and transfers between levels are determined based on a reassessment of the lowest level of input that is significant to the fair value measurement. The three levels of the fair value hierarchy are described as follows:
• Level 1 — quoted (unadjusted) market prices in active markets for identical assets or liabilities;
• Level 2 — valuation techniques for which the lowest level input that is significant to the fair value measurement is directly or indirectly observable; and
• Level 3 — valuation techniques for which the lowest level input that is significant to the fair value measurement is unobservable.
F-15
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
For recurring and non-recurring fair value measurements, external valuers may be used when internal expertise is either not available or when the valuation is deemed to be significant. External valuers are selected based on market knowledge and reputation. Where there is a significant change in fair value of an asset or liability from one period to another, an analysis is undertaken, which includes a verification of the major inputs applied in the latest valuation and a comparison, where applicable, with external sources of data.
Issued capital
Ordinary shares are classified as equity.
Incremental costs directly attributable to the issue of new shares or options are shown in equity as a deduction, net of tax, from the proceeds.
Dividends
Dividends are recognized when declared during the financial years.
Earnings/(loss) per share
Basic earnings/(loss) per share
Basic earnings/(loss) per share is calculated by dividing the profit attributable to the owners of Incannex Healthcare Limited, excluding any costs of servicing equity other than ordinary shares, by the weighted average number of ordinary shares outstanding during the financial years, adjusted for bonus elements in ordinary shares issued during the financial years. These values are set out in Note 7.
Diluted earnings/(loss) per share
Diluted earnings/(loss) per share adjusts the figures used in the determination of basic earnings per share to take into account the after income tax effect of interest and other financing costs associated with dilutive potential ordinary shares and the weighted average number of shares assumed to have been issued for no consideration in relation to dilutive potential ordinary shares. These values are set out in Note 7.
Goods and Services Tax (‘GST’) and other similar taxes
Revenues, expenses and assets are recognised net of the amount of associated GST, unless the GST incurred is not recoverable from the tax authority. In this case it is recognised as part of the cost of the acquisition of the asset or as part of the expense.
Receivables and payables are stated inclusive of the amount of GST receivable or payable. The net amount of GST recoverable from, or payable to, the tax authority is included in other receivables or other payables in the statement of financial position.
Cash flows are presented on a gross basis. The GST components of cash flows arising from investing or financing activities which are recoverable from, or payable to the tax authority, are presented as operating cash flow.
Commitments and contingencies are disclosed net of the amount of GST recoverable from, or payable to, the tax authority.
New Accounting Standards and Interpretations not yet mandatory or early adopted
International Financial Reporting Standards and Interpretations that have recently been issued or amended but are not yet mandatory, have not been early adopted by the consolidated entity for the annual reporting periods ended 30 June 2019 and 2020. The consolidated entity’s assessment of the impact of these new or amended Accounting Standards and Interpretations, most relevant to the consolidated entity, are set out below.
F-16
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
Conceptual Framework for Financial Reporting (Conceptual Framework)
The revised Conceptual Framework is applicable to annual reporting periods beginning on or after 1 January 2020 and early adoption is permitted. The Conceptual Framework contains new definition and recognition criteria as well as new guidance on measurement that affects several Accounting Standards. Where the consolidated entity has relied on the existing framework in determining its accounting policies for transactions, events or conditions that are not otherwise dealt with under the International Financial Reporting Standards, the consolidated entity may need to review such policies under the revised framework. At this time, the application of the Conceptual Framework is not expected to have a material impact on the consolidated entity’s financial statements.
IFRS 9 Financial Instruments
IFRS 9 Financial Instruments replaces IAS 39 Financial Instruments: Recognition and Measurement for the financial year ended 30 June 2019. It makes major changes to the previous guidance on the classification and measurement of financial assets and introduces an ‘expected credit loss’ model for impairment of financial assets. The investment classifications Available-for-sale financial assets and Held-to-maturity investments are no longer used and Financial assets at fair value through other comprehensive income (FVOCI) was introduced. There were no investments held in these categories as at 30 June 2018. Interest revenue is no longer included in the Revenue note and is now shown separately on the face of the statement of comprehensive income.
When adopting IFRS 9, the Group has applied transitional relief and opted not to restate prior periods due to the immaterial impact of any changes.
IFRS 15 Revenue from Contracts with Customers and Related Amending Standards
In the financial year ended 30 June 2019, the Company adopted IFRS 15 Revenue from Contracts with Customers which is effective for an annual period that begins on or after 1 January 2018. IFRS 15 introduced a 5-step approach to revenue recognition. IFRS 15 replaces IAS 18 Revenue, IAS 11 Construction Contracts and several revenue-related Interpretations. The new Standard has been applied as at 1 July 2018 using the modified retrospective approach. Under this method, the cumulative effect on initial application is recognised as an adjustment to the opening balance of accumulated losses on 1 July 2018 and comparatives are not restated. In accordance with the transition guidance, IFRS 15 has only been applied to contracts that were incomplete as at 1 July 2018. The core principle of IFRS 15 is that an entity should recognise revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. Specifically, the Standard introduces a 5-step approach to revenue recognition:
• Step 1: Identify the contract(s) with a customer.
• Step 2: Identify the performance obligations in the contract.
• Step 3: Determine the transaction price.
• Step 4: Allocate the transaction price to the performance obligations in the contract.
• Step 5: Recognise revenue when (or as) the entity satisfies a performance obligation.
In adopting IFRS 15 Steps 1 through 4 occur during the order process when the customer places the order. For on-line orders this also involves paying for the product. Step 5 occurs when the product is dispatched to the customer.
The adoption of IFRS 15 has mainly affected the following areas:
a) Sales revenue in respect of products ordered and paid for by customers upon order that had yet to be produced and delivered to the customer
F-17
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
1. Significant accounting policies (cont.)
b) The value ascribed to work-in-progress on sales that are the subject of a) above
While this represents significant new guidance, the implementation of this new guidance did not have a significant impact on the timing or amount of revenue recognised by the Company during the year ended 30 June 2019.
Financial impact of initial application of IFRS 15
First time adoption of IFRS 15 required an adjustment against Accumulated Losses of $79,220 comprising derecognition of sales revenue of $94,888 and recognition of additional cost of sales of $15,668 applicable to the prior year. For the year ended 30 June 2019, adoption of IFRS 15 had an effect of reducing net revenue by $19,111.
IFRS 17 Insurance Contracts
IFRS 17 Insurance Contracts has been issued, but is not yet mandatorily required to be adopted by the Company. The Company will be required to adopt IFRS 17 during the financial year ending 30 June 2024. The Company is not planning to early adopt this new standard and the Directors do not expect the adoption of IFRS 17 to have a material impact on the financial position or performance of the Company once adopted.
2. Critical accounting judgements, estimates and assumptions
The preparation of the financial statements requires management to make judgements, estimates and assumptions that affect the reported amounts in the financial statements. Management continually evaluates its judgements and estimates in relation to assets, liabilities, contingent liabilities, revenue and expenses. Management bases its judgements, estimates and assumptions on historical experience and on other various factors, including expectations of future events, management believes to be reasonable under the circumstances. The resulting accounting judgements and estimates will seldom equal the related actual results. The judgements, estimates and assumptions that have a significant risk of causing a material adjustment to the carrying amounts of assets and liabilities (refer to the respective notes) within the next financial year are discussed below.
Coronavirus (COVID-19) pandemic
Judgement has been exercised in considering the impacts that the Coronavirus (COVID-19) pandemic has had, or may have, on the consolidated entity based on known information. This consideration extends to the nature of the products and services offered, customers, supply chain, staffing and geographic regions in which the consolidated entity operates. Other than as addressed in specific notes, there does not currently appear to be either any significant impact upon the financial statements or any significant uncertainties with respect to events or conditions which may impact the consolidated entity unfavourably as at the reporting date or subsequently as a result of the Coronavirus (COVID-19) pandemic.
Share-based payment transactions
The consolidated entity measures the cost of equity-settled transactions with employees and third parties by reference to the fair value of the equity instruments at the date at which they are granted. The fair value is determined by using either the trinomial or Black-Scholes model taking into account the terms and conditions upon which the instruments were granted. The accounting estimates and assumptions relating to equity-settled share-based payments would have no impact on the carrying amounts of assets and liabilities within the next annual reporting period but may impact profit or loss and equity. Refer to notes 13 and 19 for further information.
F-18
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
3. Revenue & expenses
|
Consolidated |
||||
|
2020 |
2019 |
|||
|
$ |
$ |
|||
|
(a) Revenue (point in time) |
||||
|
Cannabinoid oils sales |
604,884 |
— |
||
|
604,884 |
— |
|||
|
(b) Other income |
||||
|
Income from other arrangements(1) |
123,125 |
— |
||
|
Government grants(2) |
89,500 |
— |
||
|
Interest |
4,545 |
1,553 |
||
|
217,170 |
1,553 |
|||
|
(c) Expenses |
||||
|
Executive directors’ remuneration |
539,923 |
217,949 |
||
____________
(1) Notes for Income from other contractual arrangements
In September 2018 a transaction was entered into with AXIM Biotechnologies, in consideration of the terms of the full understanding 6,800,000 IHL shares were issued in full consideration of the intended transaction.
AXIM was not able to fulfill their part of the transaction, and the contract was terminated. In lieu of returning the shares, the Company received cash. As this revenue is not derived from any normal trading transactions, it has been accounted for as a separate line item in the accounts. The return of these shares and the subsequent income is a one off income item for IHL and has not resulted in a change in equity per the consolidated statement of financial position.
(1) Notes for Government grants
Other income from government grants relates to assistance provided by the Australian Government in relation to the COVID-19 pandemic. The Company has reasonable assurance that it has complied with the conditions attaching to these grants. There were no unfulfilled conditions or other contingencies attaching to these grants as at 30 June 2020.
4. Segment Information
Identification of reportable operating segments
IFRS 8 Operating Segments requires operating segments to be identified on the basis of internal reports about components of the Group that are regularly reviewed by the Chief Executive Officer in order to allocate resources to the segment and to assess its performance.
The Group’s operating segments have been determined with reference to the monthly management accounts used by the Chief Executive Officer to make decisions regarding the Group’s operations and allocation of working capital. Due to the size and nature of the Group, the Board as a whole has been determined as the Chief Executive Officer.
Based on the quantitative thresholds included in IFRS 8, for the financial year ended 30 June 2020, the consolidated entity was organised into two operating segments based on differences in products and services provided (1) medicinal cannabis and (2) dental devices. On 30 June 2020, the Company disposed of the dental devices segment (refer note 6) to focus entirely on medicinal cannabis product sales and development from 1 July 2020. The consolidated entity will have no dental devices activities after 30 June 2020.
F-19
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
4. Segment Information (cont.)
The consolidated entity has only one geographical segment, namely Australia.
The revenues and results of these segments of the Group as a whole are set out in the condensed statement of comprehensive income and the assets and liabilities of the Group as a whole are set out in the condensed statement of financial position. A summary of revenue and expenses for the period and assets and liabilities at the end of the period for each segment is shown below.
Segment results
|
Oral and |
Medicinal Cannabis |
Unallocated |
Consolidated |
|||||||||
|
For the year ended 30 June 2020 |
|
|
|
|
||||||||
|
Revenue from external customers |
718,656 |
|
604,884 |
(1) |
— |
|
1,323,540 |
|
||||
|
Interest income |
8 |
|
2 |
|
4,543 |
|
4,553 |
|
||||
|
Other income |
140,816 |
|
212,625 |
|
— |
|
353,441 |
|
||||
|
Depreciation |
(14,854 |
) |
— |
|
— |
|
(14,854 |
) |
||||
|
Amortisation |
(21,688 |
) |
— |
|
— |
|
(21,688 |
) |
||||
|
Other expenses |
(1,591,290 |
) |
(2,899,761 |
) |
(1,851,577 |
) |
(6,342,628 |
) |
||||
|
Segment loss after income tax |
(768,352 |
) |
(2,082,250 |
) |
(1,847,034 |
) |
(4,697,636 |
) |
||||
|
Segment assets |
— |
|
662,414 |
|
3,573,665 |
|
4,236,079 |
|
||||
|
Segment liabilities |
— |
|
(567,423 |
) |
(504,228 |
) |
(1,071,651 |
) |
||||
|
|
|
|
|
|||||||||
|
For the year ended 30 June 2019 |
|
|
|
|
||||||||
|
Revenue from external customers |
1,178,466 |
|
— |
|
— |
|
1,178,466 |
|
||||
|
Interest income |
80 |
|
— |
|
1,553 |
|
1,633 |
|
||||
|
Other income |
1,800 |
|
— |
|
— |
|
1,800 |
|
||||
|
Interest expense |
— |
|
— |
|
(85,065 |
) |
(85,065 |
) |
||||
|
Depreciation |
(20,198 |
) |
— |
|
— |
|
(20,198 |
) |
||||
|
Amortisation |
(21,688 |
) |
— |
|
— |
|
(21,688 |
) |
||||
|
Other expenses |
(2,581,984 |
) |
(736,140 |
) |
(606,546 |
) |
(3,924,670 |
) |
||||
|
Income tax benefit |
151,323 |
|
— |
|
— |
|
151,323 |
|
||||
|
Segment loss after income tax |
(1,292,201 |
) |
(736,140 |
) |
(690,058 |
) |
(2,718,399 |
) |
||||
|
Segment assets |
479,553 |
|
8,237 |
|
30,121 |
|
517,911 |
|
||||
|
Segment liabilities |
(403,636 |
) |
(23,441 |
) |
(508,014 |
) |
(935,091 |
) |
||||
____________
(1) Of the total revenue from medicinal cannabis in the financial year ended 30 June 2020, 100% was through Cannvalate Pty Ltd’s distribution network.
F-20
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
5. Income tax
The prima facie income tax (expense)/benefit on pre-tax accounting (loss)/profit from operations reconciles to the income tax benefit in the financial statements as follows:
|
Consolidated |
||||||
|
2020 |
2019 |
|||||
|
$ |
$ |
|||||
|
Accounting loss before tax |
(4,697,636 |
) |
(2,869,722 |
) |
||
|
Income tax benefit at the applicable tax rate of 27.5% (2019: 27.5%) |
1,291,850 |
|
789,174 |
|
||
|
Non-deductible expenses at the applicable tax rate of 27.5% (2019:27.5%) |
(155,498 |
) |
(13,160 |
) |
||
|
Deferred tax assets not recognised |
(1,136,352 |
) |
(776,014 |
) |
||
|
Research and Development Grant in relation to prior year |
— |
|
151,323 |
|
||
|
Income tax benefit |
— |
|
151,323 |
|
||
|
|
|
|||||
|
Deductible temporary differences for which no deferred tax asset has been recognised |
|
|
||||
|
Unused tax losses at 27.5% (2019: 27.5%) |
3,872,022 |
|
2,735,670 |
|
||
|
Net unrecognised tax benefit |
3,872,022 |
|
2,735,670 |
|
||
The income tax benefit of $151,323 for the year ended 30 June 2019 is not presented on the consolidated statements of comprehensive income as it relates to income tax benefit on discontinued operations. The loss from discontinued operations is set out in note 6 to these financial statements.
The net unrecognised tax benefit has not been recognised as an asset in the financial statements because recovery of the asset is not considered probable in the context of IAS 12 Income Taxes.
The benefit will only be realised if:
a) the Company derives future assessable income of a nature and of an amount sufficient to enable the benefit to be realised.
b) the Company complies with the conditions for deductibility imposed by the law; and
c) no changes in tax legislation adversely affect the Company in realising the benefit.
6. Discontinued operations
Description
On 30 June 2020 the consolidated entity sold its 100% subsidiary — Gameday International Pty Ltd (“Gameday”), for consideration of $29,277 which was the carrying value of its assets at that date so no loss on sale was incurred. Gameday produced and sold the consolidated entity’s dental devices and had been a loss maker since 2016. As a result of the COVID-19 pandemic it suffered further as a result of the shut-down of community sport which directly affected the sale of its main product being sporting mouthguards. The sale of Gameday will allow the consolidated entity to pursue and focus entirely on its medicinal cannabis activities.
F-21
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
6. Discontinued operations (cont.)
Financial performance information
|
Consolidated |
||||||
|
2020 |
2019 |
|||||
|
$ |
$ |
|||||
|
Revenue from external customers |
718,656 |
|
1,178,466 |
|
||
|
Interest income |
8 |
|
80 |
|
||
|
Other income |
140,816 |
|
1,800 |
|
||
|
Product costs |
(589,570 |
) |
(582,209 |
) |
||
|
Administration expense |
(38,985 |
) |
(297,771 |
) |
||
|
Advertising and promotion |
(218,865 |
) |
(610,042 |
) |
||
|
Depreciation |
(14,854 |
) |
(20,198 |
) |
||
|
Amortisation |
(21,688 |
) |
(21,688 |
) |
||
|
Loss on disposal of property, plant and equipment |
(13,654 |
) |
— |
|
||
|
Impairment cost |
(82,989 |
) |
— |
|
||
|
Compliance, legal and regulatory costs |
— |
|
(27,241 |
) |
||
|
Occupancy expenses |
(81,493 |
) |
(153,830 |
) |
||
|
Salaries and employee benefit expense |
(565,734 |
) |
(910,891 |
) |
||
|
Loss before income tax |
(768,352 |
) |
(1,443,524 |
) |
||
|
Income tax benefit |
— |
|
151,323 |
|
||
|
Loss after income tax from discontinued operations |
(768,352 |
) |
(1,292,201 |
) |
||
|
|
|
|||||
|
Carrying amounts of assets and liabilities disposed |
|
|
||||
|
|
|
|||||
|
Cash |
17,970 |
|
— |
|
||
|
Inventories |
6,000 |
|
— |
|
||
|
Other current assets |
6,100 |
|
— |
|
||
|
Trade and other payables |
(793 |
) |
— |
|
||
|
Total proceeds from sale |
29,277 |
|
— |
|
||
Impairment expense
During the process of the sale of Gameday, various assets of Gameday that were unwanted by the acquirer were assessed to determine their future value or ability to be sold. Specifically, these assets included specialist or customised plant and equipment, capitalised intangible assets, and the recovery of receivables.
For each of these assets it was determined that the future value was negligible and for each the contribution to the total impairment expense is set out below:
(i) Plant and equipment
|
Original |
Accumulated Depreciation |
Book value prior |
||||||
|
76,136 |
(32,221 |
) |
43,915 |
(A) |
||||
(ii) Intangible assets
|
Original |
Accumulated Amortisation |
Book value prior |
||||||
|
116,731 |
(89,042 |
) |
27,689 |
(B) |
||||
(iii) Receivables
|
Original |
Recoverable |
Book value prior |
||||||
|
11,635 |
(250 |
) |
11,385 |
(C) |
||||
|
Impairment expense |
|
A+B+C 82,989 |
|
|||||
F-22
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
7. Loss per share
|
Basic loss per share – continuing and discontinued operations – cents |
(0.69 |
) |
(0.40 |
) |
||
|
Basic loss per share – continuing operations – cents per share |
(0.57 |
) |
(0.21 |
) |
||
|
|
|
|||||
|
Basic loss per share |
|
|
||||
|
The loss and weighted average number of ordinary shares used in the calculation of basic loss per share is as follows: |
|
|
||||
|
– Loss from continuing and discontinued operations ($) |
(4,697,636 |
) |
(2,718,399 |
) |
||
|
– Loss from continuing operations ($) |
(3,929,284 |
) |
(1,426,198 |
) |
||
|
– Weighted average number of ordinary shares (number) |
684,035,399 |
|
447,439,263 |
|
8. Dividends
The Company has not declared a dividend for the years ended 30 June 2019 or 2020.
9. Cash
|
Consolidated |
||||
|
2020 |
2019 |
|||
|
$ |
$ |
|||
|
Cash at bank and on hand |
3,603,390 |
93,332 |
||
|
3,603,390 |
93,332 |
|||
Cash at bank earns interest at floating rates based on daily bank deposit rates.
i. Reconciliation of loss for the years to net cash flows from operating activities:
|
Loss after income tax |
(4,697,636 |
) |
(2,718,399 |
) |
||
|
|
|
|||||
|
Non-cash based expenses: |
|
|
||||
|
Share based payments |
565,448 |
|
47,854 |
|
||
|
Depreciation and amortisation |
36,542 |
|
41,886 |
|
||
|
Interest expense capitalised as equity |
— |
|
75,000 |
|
||
|
Non-cash element of new business development costs |
— |
|
583,896 |
|
||
|
Other non-cash expenses |
97,221 |
|
9,413 |
|
||
|
|
|
|||||
|
Changes in net assets and liabilities: |
|
|
||||
|
(Increase)/Decrease in receivables |
(315,484 |
) |
(43,681 |
) |
||
|
(Increase)/Decrease in inventory |
(30,355 |
) |
70,268 |
|
||
|
Decrease in other current assets |
2,928 |
|
10,009 |
|
||
|
(Increase)/Decrease in trade and other payables |
464,223 |
|
(257,451 |
) |
||
|
Increase/(Decrease) in other liabilities |
(30,221 |
) |
20,772 |
|
||
|
Cash flows from (used in) operations |
(3,907,334 |
) |
(2,160,433 |
) |
ii. Non-cash financing activities
The proceeds of $29,277 from sale of the discontinued operations disclosed in note 6, were still to be received at 30 June 2020.
There Company has recorded non-cash transactions in the form of share based payments as disclosed in Note 13 to these financial statements. The total value of share-based payments recorded during the year ended 2020 is $565,448 (2019: $47,854).
F-23
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
9. Cash (cont.)
The Company recorded other current liabilities of $244,403 as at 30 June 2019, relating to option issues awaiting shareholder approval, as disclosed in note 17 to these financial statements. During the year ended 30 June 2021, this liability was settled via the issue of options upon which time the liability balance of $244,403 was transferred to equity.
10. Trade and other receivables (Current)
|
Consolidated |
||||
|
2020 |
2019 |
|||
|
$ |
$ |
|||
|
Current |
||||
|
Receivables |
276,151 |
66,605 |
||
|
GST recoverable |
137,117 |
31,179 |
||
|
413,268 |
97,784 |
|||
Opening receivables, contract assets and contract liabilities with customers:
The opening value of receivables from contracts with customers as at 1 July 2018 after the adoption of IFRS 15 was $10,422.
The opening value of contract assets from contracts with customers as at 1 July 2018 after the adoption of IFRS 15 was nil.
The opening value of contract liabilities from contracts with customers as at 1 July 2018 after the adoption of IFRS 15 was $79,220. As these opening contract liabilities related entirely to operations which have since been discontinued, no revenue is recorded in years ended 30 June 2020 and 2019 in relation to these contract liabilities. The entire value of these contract liabilities was recorded within the loss on discontinued operations, net of tax during the year ended 30 June 2019.
There was no revenue recognised in the years ended 30 June 2020 and 2019 from performance obligations satisfied (or partially satisfied) in previous periods.
Expected credit losses
The consolidated entity applies the IFRS 9 simplified model of recognising lifetime expected credit losses for all trade receivables as these items do not have a significant financing component.
In measuring the expected credit losses, the trade receivables have been assessed on a collective basis as they possess shared credit risk characteristics. They have been grouped based on the days past due and also according to the geographical location of customers.
11. Property, plant and equipment
|
Consolidated |
|||||
|
2020 |
2019 |
||||
|
$ |
$ |
||||
|
Property, plant & equipment – at cost |
— |
166,342 |
|
||
|
Less: accumulated depreciation |
— |
(80,919 |
) |
||
|
Total property, plant & equipment |
— |
85,423 |
|
||
F-24
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
11. Property, plant and equipment (cont.)
|
Reconciliation: |
Plant & Equipment |
Computer Equipment |
Office Furniture |
Total |
|||||||
|
30 June 2019 |
$ |
$ |
$ |
$ |
|||||||
|
Carrying value as at 1 July 2019 |
81,151 |
|
— |
4,272 |
|
85,423 |
|
||||
|
Disposals |
(23,065 |
) |
— |
(3,589 |
) |
(26,654 |
) |
||||
|
Depreciation |
(14,618 |
) |
— |
(236 |
) |
(14,854 |
) |
||||
|
Impairment – refer note 6(i) |
(43,468 |
) |
— |
(447 |
) |
(43,915 |
) |
||||
|
Balance at 30 June 2020 |
— |
|
— |
— |
|
— |
|
||||
|
Reconciliation: |
Plant & Equipment |
Computer Equipment |
Office Furniture |
Total |
|||||||
|
30 June 2019 |
$ |
$ |
$ |
$ |
|||||||
|
Carrying value as at 1 July 2018 |
92,339 |
|
— |
5,339 |
|
97,678 |
|
||||
|
Additions |
7,942 |
|
— |
— |
|
7,942 |
|
||||
|
Depreciation |
(19,130 |
) |
— |
(1,067 |
) |
(20,197 |
) |
||||
|
Balance at 30 June 2019 |
81,151 |
|
— |
4,272 |
|
85,423 |
|
||||
12. Other assets (current)
|
Consolidated |
||||
|
2020 |
2019 |
|||
|
$ |
$ |
|||
|
Prepayments |
11,083 |
4,683 |
||
|
Office rental bond |
25,179 |
17,179 |
||
|
Work in progress (contract assets) |
— |
17,329 |
||
|
36,262 |
39,191 |
|||
13. Share based payments
From time to time, the Company may issue equity securities (i.e. shares, options or performance rights) to its employees, directors or advisors to more closely align rewards for performance with the achievement of the Company’s growth and strategic objectives. Where the recipient is a director of the Company, shareholder approval must be sought under the ASX Listing Rules prior to the issue of any equity securities to any director.
Fair value of shares issued
The fair value of shares issued to employees is determined using the closing price of shares on the grant date and expensed over the vesting period.
Fair value of options and performance rights granted
The fair values at grant date are independently determined using either a trinomial pricing or Black-Scholes option model that take into account any price to exercise, the term of the options or rights, the share price at grant date, the price volatility of the underlying share and the risk-free interest rate for the term of the options or rights. Inputs into the trinomial and Black-Scholes option pricing models used to calculate fair value are classified as level three inputs under the fair value hierarchy of IFRS 13. The expensed fair value in the tables below represents the proportion of the total fair value that has been allocated to the current period with the balance to be expensed in future periods.
F-25
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
13. Share based payments (cont.)
The following share-based payment arrangements were put in place during the period:
|
A. Shares |
Number |
Approval |
Escrow |
Exercise Price |
Total |
Expensed |
||||||
|
Ordinary shares |
4,583,334 |
26-Jun-2020 |
n/a |
n/a |
220,000 |
220,000 |
||||||
|
Ordinary shares (escrowed) |
583,333 |
26-Jun-2020 |
30-Jun-2020 |
n/a |
28,000 |
28,000 |
||||||
|
Ordinary shares (escrowed) |
583,333 |
26-Jun-2020 |
30-Jun-2021 |
n/a |
28,000 |
304 |
||||||
|
Ordinary shares (escrowed) |
583,333 |
26-Jun-2020 |
30-Jun-2022 |
n/a |
28,000 |
152 |
||||||
|
Total shares |
6,333,333.00 |
248,456 |
|
B. Options |
Number |
Grant |
Expiry |
Exercise |
Total |
Expensed |
|||||||
|
Unlisted options |
750,000 |
26-Jun-2020 |
30-Jun-2025 |
$ |
0.05 |
24,817 |
24,817 |
||||||
|
Unlisted options |
750,000 |
26-Jun-2020 |
30-Jun-2026 |
$ |
0.05 |
26,424 |
286 |
||||||
|
Unlisted options |
750,000 |
26-Jun-2020 |
30-Jun-2027 |
$ |
0.05 |
27,754 |
151 |
||||||
|
Unlisted options |
200,000,000 |
26-Jun-2020 |
30-Sep-2021 |
$ |
0.20 |
306,299 |
130,667 |
||||||
|
Total options |
202,250,000 |
|
155,921 |
||||||||||
|
C. Performance rights |
Number |
Grant |
Expiry |
Exercise |
Total |
Expensed |
|||||||
|
Milestone-based |
2,000,000 |
26-Jun-2020 |
Various(3) |
n/a |
64,000 |
|
1,341 |
||||||
|
Value-based |
30,303,593 |
26-Jun-2020 |
24-Nov-2021 |
n/a |
811,503 |
|
184,134 |
||||||
|
Total performance rights |
32,303,593 |
|
185,475 |
||||||||||
|
Total share based payments expense(4) |
$ |
589,852 |
|||||||||||
____________
(1) These shares were issued to Directors so shareholder approval was sought and provided at a general meeting of shareholders held on 26 June 2020.
(2) Grant date is the date of the general meeting of shareholders, being 26 June 2020, at which these options and performance rights were approved by shareholders.
(3) The milestone-based performance rights have non-market milestones which must be met at various dates ranging from 31 January 2021 to 31 March 2021.
(4) The total amount issued to the Equity Reserve in relation to share based payments during the year ended 30 June 2020 per the Statement of Changes in equity is $589,852. This differs from the $565,448 disclosed in the table above by $24,404. This difference is due a $244,404 creditor that was settled via share based payments, offset by a $220,000 movement between Equity Reserve and Issued Capital.
Performance Rights
The value-based performance rights have milestones which are market-based. In arriving at the fair value of these rights the probability of achieving these milestones (related to various levels of market capitalisation) has been estimated using a trinomial option model, with major inputs being grant date share price of $0.048; risk-free rate of 0.25%; and volatility of 95%, for a total value of $469,324, of which $189,071 has been expensed in the current period commencing on 24 July 2019, being the commencement date of Dr Agarwal’s contract.
The milestone performance rights are valued at the share price at grant date ($0.048) taking into account management’s estimate s of the likelihood of meeting the milestones.
F-26
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
13. Share based payments (cont.)
Options
The fair value of the equity-settled share options granted in the above table is estimated as at the grant date using a Black-Scholes option model (for all $0.05 options) and a trinomial option model (for the $0.20 options) taking into account the terms and conditions upon which the options were granted, as follows:
|
$0.05 Options |
$0.05 Options |
$0.05 Options |
$0.20 Options |
|||||||||
|
Number |
750,000 |
|
750,000 |
|
750,000 |
|
2,000,000 |
|
||||
|
Dividend yield (%) |
0 |
% |
0 |
% |
0 |
% |
0 |
% |
||||
|
Expected volatility (%) |
92 |
% |
92 |
% |
92 |
% |
93 |
% |
||||
|
Risk-free interest rate (%) |
0.39 |
% |
0.48 |
% |
0.58 |
% |
0.25 |
% |
||||
|
Expected life of option (years) |
5 |
|
6 |
|
7 |
|
1.25 |
|
||||
|
Exercise price (cents) |
5.0 |
|
5.0 |
|
5.0 |
|
20.0 |
|
||||
|
Grant date share price (cents) |
4.8 |
|
4.8 |
|
4.8 |
|
4.8 |
|
||||
|
Vesting date |
30-Jun-2020 |
|
30-Jun-2021 |
|
30-Jun-2022 |
|
Refer (a) below |
|
||||
____________
The expected life of the options is based on historical data and is not necessarily indicative of exercise patterns that may occur. The expected volatility reflects the assumption that the historical volatility is indicative of future trends, which may also not necessarily be the actual outcome.
(a) The options vest upon the shares having a closing price of 20 cents per share or more for any 5 trading days at any time from the date of grant of the options until the expiry date of the options (30 September 2021).
Securities issued to third parties
Refer to note 19 for details of options issued to advisors and Cannvalate Pty Ltd.
14. Inventory
|
Consolidated |
||||
|
2020 |
2019 |
|||
|
$ |
$ |
|||
|
Current |
||||
|
Devices raw materials – at cost |
— |
152,804 |
||
|
Medicinal cannabis products in-transit |
183,159 |
— |
||
|
Total inventory |
183,159 |
152,804 |
||
15. Intangible assets
|
Consolidated |
||||||
|
2020 |
2019 |
|||||
|
$ |
$ |
|||||
|
Non-current |
|
|
||||
|
Trademarks & IP |
— |
|
49,377 |
|
||
|
— |
|
49,377 |
|
|||
|
Movement schedule – Trademarks & IP |
|
|
||||
|
Opening Balance |
49,377 |
|
71,066 |
|
||
|
Amortisation expense |
(21,689 |
) |
(21,689 |
) |
||
|
Impairment – refer note 6(ii) |
(27,688 |
) |
— |
|
||
|
Closing Balance |
— |
|
49,377 |
|
||
F-27
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
16. Trade and other payables (current)
|
Consolidated |
||||
|
2020 |
2019 |
|||
|
$ |
$ |
|||
|
Trade payables |
590,099 |
376,124 |
||
|
Accrued expenses |
316,046 |
65,797 |
||
|
Employee leave entitlements |
48,861 |
36,899 |
||
|
955,006 |
478,820 |
|||
|
Employee leave entitlements Reconciliation: |
Consolidated |
|
|
$ |
||
|
Year ended 30 June 2020 |
||
|
Carrying value as at 1 July 2019 |
36,899 |
|
|
Leave accrued by employees during the year |
11,962 |
|
|
Balance at 30 June 2020 |
48,861 |
|
$ |
|||
|
Year ended 30 June 2019 |
|
||
|
Carrying value as at 1 July 2018 |
45,786 |
|
|
|
Leave used by employees during the year |
(8,887 |
) |
|
|
Balance at 30 June 2019 |
36,899 |
|
|
17. Other current liabilities
|
Income received in advance(1) |
— |
146,868 |
||
|
Provision for sales refunds(1) |
116,645 |
— |
||
|
Options issues awaiting shareholder approval(2) |
— |
244,403 |
||
|
116,645 |
391,271 |
____________
(1) Under the terms of the sale agreement for the disposal of the devices business (refer to note 6) the Company is liable to pay to the buyer of the devices business the value of any sales proceeds already received by the Company where devices will be delivered to the customer by the buyer of the devices business after 30 June 2020. In prior years, this item related to sales proceeds that had been received where the device had yet to be produced and shipped to the customer and was treated under IFRS15 as income received in advance.
(2) On 9 August 2019, at a general meeting of shareholders, the issue of 120,000,000 options to Cannvalate Pty Ltd as remuneration for Cannvalate’s management of the Company’s clinical program was approved. This amount was transferred to the equity based premium reserve upon approval (refer also to note 19).
|
Provision for sales refunds Reconciliation: |
Consolidated |
|
|
$ |
||
|
Year ended 30 June 2020 |
||
|
Carrying value as at 1 July 2019 |
— |
|
|
Transfer from income received in advance |
116,645 |
|
|
Balance at 30 June 2020 |
116,645 |
There was no opening or closing balance of the provision for sales refunds in the year ended 30 June 2019.
F-28
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
18. Issued capital
(a) Issued Capital
|
34,192,043 |
26,951,744 |
(b) Ordinary shares — movements during years
|
Year ended |
Year ended |
|||
|
At beginning of year |
581,897,040 |
288,288,248 |
||
|
Issues of new shares – placements |
114,663,460 |
195,203,398 |
||
|
Issues of new shares – rights issues |
— |
73,572,062 |
||
|
Issues of new shares – share based payments |
5,750,000 |
— |
||
|
Conversion of performance rights |
11,916,668 |
24,833,332 |
||
|
Exercise of options |
34,427,321 |
— |
||
|
At end of year |
748,654,489 |
581,897,040 |
Ordinary shares entitle the holder to participate in dividends and the proceeds on winding up of the Company in proportion to the number of shares held. On a show of hands, every shareholder present at a meeting is entitled to one vote and upon a poll each share is entitled to one vote. Ordinary shares have no par value and the Company does not have a limited amount of authorised capital.
(c) Movement in number of options on issue for the years
At 30 June 2020
|
Expiry date and exercise price |
Balance at |
Granted |
Exercised/ (expired) |
Balance at |
|||||||||
|
30-Sep-2020 $0.04 IHLOB |
|
262,960,728 |
|
— |
|
(2,427,321 |
) |
|
260,533,407 |
||||
|
01-Jan-2020 $0.02 unlisted(1) |
|
— |
|
10,000,000 |
|
(10,000,000 |
) |
|
— |
||||
|
01-May-2020 $0.03 unlisted(1) |
|
— |
|
10,000,000 |
|
(10,000,000 |
) |
|
— |
||||
|
01-May-2020 $0.04 unlisted(1) |
|
— |
|
12,000,000 |
|
(12,000,000 |
) |
|
— |
||||
|
01-Dec-2020 $0.06 unlisted(1) |
|
— |
|
14,000,000 |
|
— |
|
|
14,000,000 |
||||
|
01-Dec-2020 $0.08 unlisted(1) |
|
— |
|
16,000,000 |
|
— |
|
|
16,000,000 |
||||
|
01-Dec-2020 $0.10 unlisted(1) |
|
— |
|
18,000,000 |
|
— |
|
|
18,000,000 |
||||
|
01-Dec-2020 $0.12 unlisted(1) |
|
— |
|
20,000,000 |
|
— |
|
|
20,000,000 |
||||
|
01-Dec-2020 $0.14 unlisted(1) |
|
— |
|
20,000,000 |
|
— |
|
|
20,000,000 |
||||
|
30-Sep-2021 $0.08 unlisted(2) |
|
— |
|
89,919,705 |
|
— |
|
|
89,919,705 |
||||
|
30-Sep-2021 $0.20 unlisted(3) |
|
— |
|
200,000,000 |
|
— |
|
|
200,000,000 |
||||
|
30-Jun-2025 $0.05 unlisted(4) |
|
— |
|
750,000 |
|
— |
|
|
750,000 |
||||
|
30-Jun-2026 $0.05 unlisted(4) |
|
— |
|
750,000 |
|
— |
|
|
750,000 |
||||
|
30-Jun-2027 $0.05 unlisted(4) |
|
— |
|
750,000 |
|
— |
|
|
750,000 |
||||
|
Total |
|
262,960,728 |
|
412,169,705 |
|
(34,427,321 |
) |
|
640,703,112 |
||||
|
Weighted average price ($) |
$ |
0.04 |
$ |
0.139 |
$ |
0.031 |
|
$ |
0.104 |
||||
F-29
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
18. Issued capital (cont.)
At 30 June 2019
|
Expiry date and exercise price |
Balance at |
Granted |
Expired |
Balance at |
|||||||||
|
31-Dec-2019 $0.12 (IHLOA) |
|
17,266,857 |
|
— |
|
(17,266,857 |
) |
|
— |
||||
|
31-Dec-2019 $0.12 unlisted |
|
11,750,000 |
|
— |
|
(11,750,000 |
) |
|
— |
||||
|
31-Dec-2019 $0.128 unlisted |
|
1,171,879 |
|
— |
|
(1,171,879 |
) |
|
— |
||||
|
30-Sep-2020 $0.04 IHLOB |
|
126,570,156 |
|
136,390,572 |
|
— |
|
|
262,960,728 |
||||
|
Total |
|
156,758,892 |
|
136,390,572 |
|
(30,188,736 |
) |
|
262,960,728 |
||||
|
Weighted average price ($) |
$ |
0.055 |
$ |
0.040 |
$ |
0.120 |
|
$ |
0.040 |
||||
____________
(1) A total of 120,000,000 options were issued to Cannvalate Pty Ltd upon approval by shareholders on 9 August 2019.
(2) 22,368,422 options were issued to participants of the July 2019 equity capital raisings attaching to shares subscribed for under those raisings and 33,000,000 options were issued to brokers who supported those equity capital raisings. A further 34,551,283 options were issued to participants of the October 2019 capital raising attaching to shares subscribed for under that raising.
(3) 200,000,000 options were issued as remuneration for the Company’s Chief Medical Officer (Dr Sud Agarwal), after approval by shareholders on 26 June 2020.
(4) 2,250,000 options were issued as remuneration for the Company’s Chief Executive Officer (Mr Joel Latham), after approval by shareholders on 26 June 2020.
(d) Movement in number of Performance Shares and Performance Rights for the years
At 30 June 2020
|
Security Description |
Balance at |
Granted by the Company |
Converted or Expired |
Balance at |
|||||
|
Performance Rights(1) |
24,166,668 |
32,303,593 |
(14,916,668 |
) |
41,553,593 |
||||
|
Performance Shares(2) |
20,000,002 |
— |
(20,000,002 |
) |
— |
||||
At 30 June 2019
|
Security Description |
Balance at |
Granted by the Company |
Converted or Expired |
Balance at |
|||||
|
Performance Rights |
735,021 |
49,000,000 |
(25,568,353 |
) |
24,166,668 |
||||
|
Performance Shares |
40,000,004 |
— |
(20,000,002 |
) |
20,000,002 |
||||
____________
(1) 32,303,593 performance rights were issued as remuneration for the Company’s Chief Medical Officer (Dr Sud Agarwal), after approval by shareholders on 26 June 2020. 11,916,668 performance rights converted into ordinary shares upon achievement of designated performance hurdles and 3,000,000 performance rights expired.
(2) Performance shares were issued to holders upon the Company’s relisting in November 2016. Performance hurdles attaching to these shares related to sales targets within the now discontinued devices business. These targets were not achieved and the performance shares lapsed on 30 June 2020.
F-30
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
19. Reserves
Equity based premium reserve
|
Consolidated |
||||
|
2020 |
2019 |
|||
|
$ |
$ |
|||
|
Balance at start of year |
451,643 |
228,725 |
||
|
Options issued to advisors(1) |
449,093 |
175,064 |
||
|
Options issued to Cannvalate Pty Ltd(2) |
244,403 |
— |
||
|
Equity instruments issued to management and directors |
345,449 |
47,854 |
||
|
Balance at end of year |
1,490,588 |
451,643 |
||
____________
(1) During the year ended 30 June 2020, 33,000,000 options exercisable at $0.08 and expiring on 30 September 2021, were issued to brokers who supported the July 2019 capital raisings. These options have been valued using a Black-Scholes option model with inputs being grant date share price of $0.04 risk-free rate of 0.24% and volatility of 92%.
(2) On 9 August 2019, at a general meeting of shareholders, the issue of 120,000,000 options to Cannvalate Pty Ltd as remuneration for Cannvalate’s management of the Company’s clinical program was approved. This amount was initially recorded as a payable as at 30 June 2019 (refer also to note 17) and transferred to the reserve in the year ended 30 June 2020. Details of these options are set out in note 18(c) and have been valued using Black-Scholes option model with inputs being grant date share price of $0.02; risk-free rate of 1.07% and volatility of 59%.
The equity based premium reserve is used to record the value of equity issued to raise capital, and for share-based payments.
20. Remuneration of auditors
|
Consolidated |
||||
|
2020 |
2019 |
|||
|
$ |
$ |
|||
|
Audit or review of the financial reports of the Company |
||||
|
Amounts received & receivable by the auditor: |
||||
|
Audit services – HLB Mann Judd |
37,000 |
37,500 |
||
|
37,000 |
37,500 |
|||
The above remuneration of auditors has been recorded within Administration expense in the statement of comprehensive income/(loss).
21. Financial Instruments
The Group’s principal financial instruments comprise cash and short-term deposits and convertible notes.
The main purpose of these financial instruments is to raise finance for the Group’s operations. The Group has various other financial liabilities such as trade payables, which arise directly from its operations. It is, and has been throughout the years, the Group’s policy that no trading in financial instruments shall be undertaken. The main risks arising from the Group’s financial instruments are cash flow interest rate risk, liquidity risk, and credit risk. The Board reviews and agrees policies for managing each of these risks and they are summarised below.
Details of the significant accounting policies and methods adopted, including the criteria for recognition, the basis of measurement and the basis on which income and expenses are recognised, in respect of each class of financial asset, financial liability and equity instrument are disclosed in note 1 to the financial statements.
F-31
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
21. Financial Instruments (cont.)
(a) Interest rate risk
The Group’s exposure to the risk of changes in market interest rates relates primarily to the Group’s short-term deposits with a floating interest rate.
The Group’s exposure to interest rate on financial assets and financial liabilities is detailed in the sensitivity analysis section of this note.
(b) Sensitivity analysis
During the years ended 30 June 2019 and 2020, if interest rates had been 50 basis points higher or lower than the prevailing rates realised, with all other variables held constant, there would have been an immaterial change in post-tax result for the year. The impact on equity would have been the same.
(c) Net fair values
The net fair value of cash and non-interest bearing monetary financial assets and liabilities approximates their carrying value.
(d) Commodity price risk
The Group’s exposure to price risk is minimal.
(e) Credit risk
There are no significant concentrations of credit risk within the Group.
With respect to credit risk arising from the other financial assets of the Group, which comprise cash, available-for-sale financial assets and certain derivative instruments, the Group’s exposure to credit risk arises from default of the counter party, with a maximum exposure equal to the carrying amount of these instruments.
Since the Group trades only with recognised third parties, there is no requirement for collateral.
(f) Liquidity risk
The Group’s objective is to maintain a balance between continuity of funding and flexibility through the use of share issues and convertible notes.
The Group’s contractual liabilities at 30 June 2020 were as follows:
|
Description |
Less than |
1 to 3 |
3 months to |
1 to 5 |
Total |
|||||
|
$ |
$ |
$ |
$ |
$ |
||||||
|
Consolidated |
||||||||||
|
Payables & accruals |
906,144 |
— |
— |
— |
906,144 |
|||||
|
906,144 |
— |
— |
— |
906,144 |
The Group’s contractual liabilities at 30 June 2019 were as follows:
|
Description |
Less than |
1 to 3 |
3 months to |
1 to 5 |
Total |
|||||
|
$ |
$ |
$ |
$ |
$ |
||||||
|
Consolidated |
||||||||||
|
Payables & accruals |
478,820 |
— |
— |
— |
478,820 |
|||||
|
Borrowings |
65,000 |
— |
— |
— |
65,000 |
|||||
|
543,820 |
— |
— |
— |
543,820 |
F-32
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
21. Financial Instruments (cont.)
(g) Capital Management
The Group’s objectives when managing capital are to safeguard its ability to continue as a going concern, so that it may continue to provide returns for shareholders and benefits for other stakeholders. Due to the nature of the Group’s past activities, being a drug development business , it does not have ready access to credit facilities and therefore is not subject to any externally imposed capital requirements, with the primary source of Group funding being equity raisings and unsecured convertible notes. Accordingly, the objective of the Group’s capital risk management is to balance the current working capital position against the requirements and corporate overheads. This is achieved by maintaining appropriate liquidity to meet anticipated operating requirements, with a view to initiating fund raisings as required.
22. Commitments and contingencies
Lease commitments
The Group holds two commercial leases for its office premises in Melbourne and Sydney, Australia. Both of these leases had terms of 12 months from the commencement date of the lease. Future minimum payments under these contracts as at 30 June are as follows:
|
Consolidated |
||||
|
2020 |
2019 |
|||
|
$ |
$ |
|||
|
Within one year |
9,697 |
11,500 |
||
|
Total minimum contract payments |
9,697 |
11,500 |
||
In transitioning to IFRS 16, these leases were not capitalised on the basis that these are short-term leases.
23. Key Management Personnel compensation and related party disclosure
The Key Management Personnel of Incannex Healthcare Limited during the years were:
Troy Valentine
Peter Widdows
Joel Latham
Sud Agarwal (commenced 24 July 2019)
Alistair Blake (ceased as a director on 24 July 2019 and ceased employment on 31 October 2019)
Key management personnel compensation
|
Consolidated |
||||
|
2020 |
2019 |
|||
|
$ |
$ |
|||
|
Short-term employee benefits |
638,201 |
447,929 |
||
|
Long-term employment benefits |
565,448 |
42,818 |
||
|
Post-employment benefits |
29,985 |
14,344 |
||
|
Total KMP compensation |
1,233,634 |
505,091 |
||
Transactions with related entities
Transactions between related parties are on commercial terms and conditions, no more favourable than those available to other parties unless otherwise stated.
F-33
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
23. Key Management Personnel compensation and related party disclosure (cont.)
During the year ended 30 June 2020, $145,200 (2019: $115,864) fees were paid to Alignment Capital Pty Ltd (“Alignment”), an entity in which Mr Valentine is a director. Alignment was engaged by the Company to act as lead manager in the various capital raisings conducted during the year.
As at 30 June 2019, there was $50,000 payable to Alignment Capital Pty Ltd and $15,000 payable to Joel Latham as short-term loans. These loans were repaid on 15 July 2019. There were no other amounts due to related parties as at 30 June 2019.
Cannvalate Pty Ltd (Cannvalate) is an entity of which Dr Sud Agarwal is a significant shareholder, the CEO and a director. In March 2019, the Company entered into a distribution agreement with Cannvalate. As stated in Note 4, of the total revenue from medicinal cannabis in the financial year ended 30 June 2020, 100% was through Cannvalate’s distribution network. This agreement is no longer effective and was terminated in June 2021.
As stated in Note 19, On 9 August 2019, at a general meeting of shareholders, the issue of 120,000,000 options to Cannvalate as remuneration for Cannvalate’s management of the Company’s clinical program was approved. This amount was initially recorded as a payable as at 30 June 2019 (refer also to Note 17) and transferred to reserves in the year ended 30 June 2020.
There were no amounts payable to related parties as at 30 June 2020.
24. Details of the controlled entity
The consolidated financial statements include the financial statements of Incannex Healthcare Limited (‘IHL’) and its wholly owned subsidiary Incannex Pty Ltd (‘IXPL’). IXPL is incorporated in Australia and IHL owns 100% of the issued ordinary shares in IXPL (2019: 100%).
On 30 June 2020, the consolidated entity disposed entirely of its 100% subsidiary — Gameday International Pty Ltd, (‘Gameday’). As at 30 June 2019, the consolidated entity owned 100% of the issued ordinary shares of Gameday.
25. Subsequent events
Between 30 June 2020 and the date these financial statements were authorized for issue by the Board of Directors (17 August 2021), holders of options have provided $584,290 to exercise a total of 14,607,242 ‘IHLOB’ options into IHL ordinary shares.
Between 30 June 2020 and the date these financial statements were authorized for issue, the Group has issued the following securities:
a. 2,952,619 ordinary shares issued on 1 July 2020, granted via three tranches of 984,207 shares each. Each of these tranches are subject to escrow restrictions expiring on 30 June 2021, 30 June 2022 and 30 June 2023 respectively;
b. 2,250,000 unlisted options issued on 1 July 2020 with an exercise price of $0.05, granted via three tranches of 750,000 options each. These three tranches are subject to vesting dates of 30 June 2025, 30 June 2026 and 30 June 2027 respectively;
c. 30,164,690 unlisted options issued on 2 October 2020 with an exercise price of $0.08 and a vesting date of 30 September 2021; and
d. 20,000,000 unlisted options issued on 20 November 2020. These options were granted via two tranches of 10,000,000 options each, one tranche with an exercise price of $0.15 and the other with an exercise price of $0.25. Both of these tranches have a vesting date of 20 November 2024.
There have been no other material events subsequent to 30 June 2020.
F-34
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
For the years ended 30 June 2019 and 2020
26. Parent entity disclosures
Incannex Healthcare Limited (ACN 096 635 246) is the parent entity which is registered and domiciled in Australia.
The registered address of the parent entity is Level 39, Rialto Tower South, 525 Collins Street, Melbourne, Victoria, Australia.
The individual financial statements for the parent entity show the following aggregate amounts. The information presented has been prepared using accounting policies as discussed in Note 1.
|
2020 |
2019 |
|||||
|
$ |
$ |
|||||
|
Financial Position as at 30 June 2020 and 2019 |
|
|
||||
|
Current assets |
3,573,665 |
|
30,120 |
|
||
|
Non-Current assets(i) |
— |
|
7,383,665 |
|
||
|
Total assets |
3,573,665 |
|
7,413,785 |
|
||
|
Current liabilities |
(504,228 |
) |
(508,014 |
) |
||
|
Non-current liabilities |
— |
|
— |
|
||
|
Total liabilities |
(504,228 |
) |
(508,014 |
) |
||
|
Net assets |
3,069,437 |
|
6,905,771 |
|
||
|
|
|
|||||
|
Issued capital |
34,192,043 |
|
26,951,744 |
|
||
|
Reserves |
1,490,588 |
|
451,643 |
|
||
|
Accumulated losses |
(32,613,194 |
) |
(20,497,616 |
) |
||
|
Shareholders’ equity |
3,069,437 |
|
6,905,771 |
|
||
____________
(i) In the year ended 30 June 2020, the loan to the subsidiary company has been fully impaired.
Contingencies of the Parent Entity
There were no contingent liabilities involving the parent entity as at 30 June 2020 (2019: Nil).
Guarantees of the Parent Entity
There were no guarantees involving the parent entity as at 30 June 2020 (2019: Nil)
F-35
Condensed Consolidated Statements of Comprehensive Income
for the Half-Years ended 31 December 2020 and 2019 (Unaudited)
|
Note |
31 December 2020 |
31 December 2019 |
||||||
|
$ |
$ |
|||||||
|
Sales |
2(a) |
1,177,163 |
|
7,350 |
|
|||
|
Product costs |
(537,939 |
) |
(8,450 |
) |
||||
|
639,224 |
|
(1,100 |
) |
|||||
|
Other income |
2(b) |
52,078 |
|
2,929 |
|
|||
|
Administration expenses |
(454,664 |
) |
(173,228 |
) |
||||
|
Advertising and investor relations |
(227,532 |
) |
(141,783 |
) |
||||
|
Compliance, legal and regulatory |
(89,065 |
) |
(56,270 |
) |
||||
|
Research and development costs |
(2,039,147 |
) |
(313,426 |
) |
||||
|
Share based payment expense |
5 |
(380,371 |
) |
(966,937 |
) |
|||
|
Occupancy expenses |
(61,992 |
) |
(1,042 |
) |
||||
|
Salaries and employee benefit expense |
(327,920 |
) |
(274,616 |
) |
||||
|
Loss before tax from continuing operations |
(2,889,389 |
) |
(1,925,473 |
) |
||||
|
Income tax benefit (expense) |
— |
|
— |
|
||||
|
Loss after tax from continuing operations |
(2,889,389 |
) |
(1,925,473 |
) |
||||
|
Loss after tax from discontinuing operations |
12 |
— |
|
(286,531 |
) |
|||
|
Net loss for the period |
(2,889,389 |
) |
(2,212,004 |
) |
||||
|
Other comprehensive income |
— |
|
— |
|
||||
|
Total comprehensive loss for the period |
(2,889,389 |
) |
(2,212,004 |
) |
||||
|
Total comprehensive loss attributable to owners of the parent |
(2,889,389 |
) |
(2,212,004 |
) |
||||
|
|
|
|||||||
|
Earnings per share from continuing operations |
3 |
|
|
|||||
|
Basic loss per share (cents per share) |
(0.32 |
) |
(0.30 |
) |
||||
|
Diluted loss per share (cents per share) |
(0.32 |
) |
(0.30 |
) |
||||
|
Earnings per share from discontinued operations |
3 |
|
|
|||||
|
Basic loss per share (cents per share) |
— |
|
(0.04 |
) |
||||
|
Diluted loss per share (cents per share) |
— |
|
(0.04 |
) |
||||
The accompanying notes form part of these condensed consolidated financial statements
F-36
Condensed Consolidated Statements of Financial Position
as at 31 December 2020 and 30 June 2020 (Unaudited)
|
Note |
31 December 2020 |
30 June |
||||||
|
$ |
$ |
|||||||
|
Assets |
|
|
||||||
|
Current assets |
|
|
||||||
|
Cash |
11,840,308 |
|
3,603,390 |
|
||||
|
Trade and other receivables |
87,754 |
|
413,268 |
|
||||
|
Other financial assets |
39,963 |
|
36,262 |
|
||||
|
Inventory |
212,927 |
|
183,159 |
|
||||
|
Total current assets |
12,180,952 |
|
4,236,079 |
|
||||
|
|
|
|||||||
|
Total assets |
12,180,952 |
|
4,236,079 |
|
||||
|
|
|
|||||||
|
Liabilities |
|
|
||||||
|
Current liabilities |
|
|
||||||
|
Trade and other payables |
378,449 |
|
955,006 |
|
||||
|
Other current liabilities |
— |
|
116,645 |
|
||||
|
Total current liabilities |
378,449 |
|
1,071,651 |
|
||||
|
|
|
|||||||
|
Total liabilities |
378,449 |
|
1,071,651 |
|
||||
|
Net assets |
11,802,503 |
|
3,164,428 |
|
||||
|
|
|
|||||||
|
Equity |
|
|
||||||
|
Issued capital |
7 |
45,076,484 |
|
34,192,043 |
|
|||
|
Reserves |
2,133,611 |
|
1,490,588 |
|
||||
|
Accumulated losses |
(35,407,592 |
) |
(32,518,203 |
) |
||||
|
Total equity |
11,802,503 |
|
3,164,428 |
|
||||
The accompanying notes form part of these condensed consolidated financial statements
F-37
Condensed Consolidated Statements of Cash Flows
for the Half-Years ended 31 December 2020 and 2019 (Unaudited)
|
Consolidated |
||||||
|
31 December 2020 |
31 December 2019 |
|||||
|
$ |
$ |
|||||
|
Cash flows from operating activities |
|
|
||||
|
Receipts from customers |
1,288,845 |
|
351,594 |
|
||
|
Payment to suppliers and employees |
(4,235,483 |
) |
(2,022,124 |
) |
||
|
Interest and other income received |
52,078 |
|
143,747 |
|
||
|
Net cash used in operating activities |
(2,894,560 |
) |
(1,526,783 |
) |
||
|
|
|
|||||
|
Cash flows from investing activities |
|
|
||||
|
Proceeds from sale of discontinued operations |
29,276 |
|
— |
|
||
|
Net cash provided by financing activities |
29,276 |
|
— |
|
||
|
|
|
|||||
|
Cash flows from financing activities |
|
|
||||
|
Proceeds from share issues |
11,200,178 |
|
7,119,901 |
|
||
|
Share issue costs paid |
(97,975 |
) |
(493,385 |
) |
||
|
Repayment of debt |
— |
|
(65,000 |
) |
||
|
Net cash provided by financing activities |
11,102,203 |
|
6,561,516 |
|
||
|
|
|
|||||
|
Net increase in cash |
8,236,918 |
|
5,034,733 |
|
||
|
Cash at beginning of period |
3,603,390 |
|
93,332 |
|
||
|
Cash at end of period |
11,840,308 |
|
5,128,065 |
|
||
The accompanying notes form part of these condensed consolidated financial statements
F-38
Condensed Consolidated Statements of Changes in Equity
for the Half-Years ended 31 December 2020 and 2019 (Unaudited)
|
Consolidated |
Issued |
Reserves |
Accumulated |
Total |
|||||||
|
$ |
$ |
$ |
$ |
||||||||
|
Balance at 1 July 2019 |
26,951,744 |
|
451,643 |
(27,820,567 |
) |
(417,180 |
) |
||||
|
Loss for the period |
— |
|
— |
(2,212,004 |
) |
(2,212,004 |
) |
||||
|
Other comprehensive income |
— |
|
— |
— |
|
— |
|
||||
|
Total comprehensive loss for the period |
— |
|
— |
(2,212,004 |
) |
(2,212,004 |
) |
||||
|
Placement shares issued |
6,885,200 |
|
— |
— |
|
6,885,200 |
|
||||
|
Shares issued on exercise of options |
234,216 |
|
— |
— |
|
234,216 |
|
||||
|
Options granted |
— |
|
244,734 |
— |
|
244,734 |
|
||||
|
Share based payments |
— |
|
966,937 |
— |
|
966,937 |
|
||||
|
Shares issued pursuant to prospectus |
154 |
|
— |
— |
|
154 |
|
||||
|
Share issue costs |
(493,384 |
) |
— |
— |
|
(493,384 |
) |
||||
|
Balance at 31 December 2019 |
33,577,930 |
|
1,663,314 |
(30,032,571 |
) |
5,208,673 |
|
||||
|
Consolidated |
Issued |
Reserves |
Accumulated |
Total |
|||||||
|
$ |
$ |
$ |
$ |
||||||||
|
Balance at 1 July 2020 |
34,192,043 |
|
1,490,588 |
(32,518,203 |
) |
3,164,428 |
|
||||
|
Loss for the period |
— |
|
— |
(2,889,389 |
) |
(2,889,389 |
) |
||||
|
Other comprehensive income |
— |
|
— |
— |
|
— |
|
||||
|
Total comprehensive loss for the period |
— |
|
— |
(2,889,389 |
) |
(2,889,389 |
) |
||||
|
Shares issued on exercise of options |
11,199,678 |
|
— |
— |
|
11,199,678 |
|
||||
|
Options granted |
— |
|
262,652 |
— |
|
262,652 |
|
||||
|
Share based payments |
— |
|
380,371 |
— |
|
380,371 |
|
||||
|
Share issue costs |
(315,237 |
) |
— |
— |
|
(315,237 |
) |
||||
|
Balance at 31 December 2020 |
45,076,484 |
|
2,133,611 |
(35,407,592 |
) |
11,802,503 |
|
||||
The accompanying notes form part of these condensed consolidated financial statements
F-39
Notes to the Condensed Consolidated Financial Statements
for the Half-Years ended 31 December 2020 and 2019
NOTE 1: CONDENSED STATEMENT OF SIGNIFICANT ACCOUNTING POLICIES
(a) Basis of preparation
The condensed interim consolidated financial statements (the interim financial statements) are general purpose interim financial statements and have been have prepared in accordance with the requirements of the Corporations Act 2001, applicable accounting standards including IAS 34 Interim Financial Reporting, Accounting Interpretations and other authoritative pronouncements of the International Accounting Standards Board (IASB). Compliance with IFRS 134 ensures compliance with IAS 34 ‘Interim Financial Reporting’.
The interim financial statements comprise the condensed interim financial statements for Incannex Healthcare Limited (the “Company”) and its consolidated subsidiaries (collectively, the “Group”). For the purposes of preparing the interim financial statements, the Group is a for-profit entity.
The interim financial statements do not include full disclosures of the type normally included in the full financial report. Therefore, it cannot be expected to provide as full an understanding of the financial performance, financial position and cash flows of the Group as in the full financial report. It is recommended interim financial statements be read in conjunction with the full financial report for the years ended 30 June 2020 and 2019 and any public announcements made by Incannex Healthcare Limited and its subsidiaries during the half-years in accordance with continuous disclosure requirements arising under the Corporations Act 2001 and the ASX Listing Rules.
The accounting policies and methods of computation adopted are consistent with those of the previous financial year and corresponding half-year except for the impact of the new standards and interpretations effective 1 July 2020 as outlined below. These accounting policies are consistent with International Financial Reporting Standards and with International Financial Reporting Standards. To ensure comparability with current year disclosures, some presentation changes have been made to comparative information.
The interim financial statements have been prepared on a historical cost basis, except for the revaluation of certain financial instruments to fair value. Cost is based on the fair value of the consideration given in exchange for assets.
The Group is domiciled in Australia and all amounts are presented in Australian dollars, unless otherwise noted.
For the purpose of preparing the interim financial statements, the half-year has been treated as a discrete reporting period.
(b) Adoption of new and revised standards
New Standards and Interpretations applicable for the half years ended 31 December 2020 and 2019
In the half years ended 31 December 2020 and 2019, the Directors have reviewed all of the new and revised Standards and Interpretations issued by the IFRS that are relevant to the Group and effective for the half-years. As a result of this review, the Directors have determined that there is no material impact of the new and revised Standards and Interpretations on the Group and, therefore, no material change is necessary to Group accounting policies.
Standards and Interpretations in issue not yet adopted
The Directors have also reviewed all of the new and revised Standards and Interpretations in issue not yet adopted for the half-year ended 31 December 2020. As a result of this review the Directors have determined that there is no material impact of the Standards and Interpretations in issue not yet adopted on the Group and, therefore, no change is necessary to Group accounting policies.
IFRS 17 Insurance Contracts
IFRS 17 Insurance Contracts has been issued, but is not yet mandatorily required to be adopted by the Company. The Company will be required to adopt IFRS 17 during the financial year ending 30 June 2024. The Company is not planning to early adopt this new standard and the Directors do not expect the adoption of IFRS 17 to have a material impact on the financial position or performance of the Company once adopted.
F-40
Notes to the Condensed Consolidated Financial Statements
for the Half-Years ended 31 December 2020 and 2019
NOTE 1: CONDENSED STATEMENT OF SIGNIFICANT ACCOUNTING POLICIES (cont.)
(c) Statement of compliance
The interim financial statements were authorised for issue on 2 August 2021 by the Board of Directors.
The interim financial statements comply with International Financial Reporting Standards (IFRS), as issued by the International Accounting Standards Board (IASB).
(d) Significant accounting estimates and judgements
The preparation of the interim financial statements requires management to make judgments, estimates and assumptions that affect the application of accounting policies and the reported amounts of assets, liabilities, income and expense. Actual results may differ from these estimates.
The judgements, estimates and assumptions applied in the interim financial statements, including the key sources of estimation uncertainty were the same as those applied in the Group’s last annual financial statements for the year ended 30 June 2020.
(e) Going concern
The financial report has been prepared on the going concern basis, which contemplates continuity of normal business activities and the realisation of assets and settlements of liabilities in the ordinary course of business.
NOTE 2: REVENUE AND OTHER INCOME
(a) Revenue from contracts with customers
The Group derives its revenue from the sale of medicinal cannabinoid oils.
This is consistent with the revenue information that is disclosed for each reportable segment under IFRS 8 (see note 4).
|
for the Half-Years ended 31 December 2020 and 2019 (Unaudited) |
31 December |
31 December |
||
|
Point in time: |
||||
|
Sales of cannabinoid oils |
1,177,163 |
7,350 |
||
|
Total sales revenue |
1,177,163 |
7,350 |
(b) Other income
|
Income from other contractual arrangements |
50,171 |
— |
||
|
Interest |
1,907 |
2,929 |
||
|
Total other income |
52,078 |
2,929 |
The Company disaggregates revenue from contracts with customers based on the categories that most closely depict how the nature, amount, timing and uncertainty of revenue and cash flows are affected by economic factors. During the half-years ended 31 December 2020 and 2019, the Company recognized revenue from only one such category, being cannabinoid oils sales. The Company previously recognized revenue from oral and dental devices, although these operations have been discontinued. All sales are made within Australia and the Company has not disaggregated revenue based on geography.
F-41
Notes to the Condensed Consolidated Financial Statements
for the Half-Years ended 31 December 2020 and 2019
NOTE 3: LOSS PER SHARE
Basic loss per share has been calculated using the loss attributable to shareholders of the Parent Company and the weighted average number of ordinary shares on issue.
|
for the Half-Years ended 31 December 2020 and 2019 (Unaudited) |
31 December |
31 December |
||
|
Weighted average number of shares |
902,054,732 |
649,048,889 |
NOTE 4: SEGMENT REPORTING
IFRS 8 Operating Segments requires operating segments to be identified on the basis of internal reports about components of the Group that are regularly reviewed by the Chief Executive Officer in order to allocate resources to the segment and to assess its performance.
The Group’s operating segments have been determined with reference to the monthly management accounts used by the Chief Executive Officer to make decisions regarding the Group’s operations and allocation of working capital. Due to the size and nature of the Group, the Board as a whole has been determined as the Chief Executive Officer.
Based on the quantitative thresholds included in IFRS 8, for the half-years, the Group now has two reportable segments, being (1) distribution of medicinal cannabis products; and (2), development of psychedelic medicines and therapies — the latter commenced during the half-year ended 31 December 2020 — and currently one geographical segment, namely Australia.
The revenues and results of these segments of the Group as a whole are set out in the condensed consolidated statement of comprehensive income and the assets and liabilities of the Group as a whole are set out in the condensed consolidated statement of financial position.
In the corresponding period for FY20, the Group had two reportable segments, being (1) production and distribution of dental devices; and (2) distribution of medicinal cannabis products. The production and distribution of dental devices ceased on 30 June 2020 and the results for this segment are reported in the condensed consolidated statement of comprehensive income as ‘Loss after tax on discontinued operations’.
A summary of revenue and expenses for the half-years and assets and liabilities at the end of the half-years for each segment is shown below:
|
6 months ended 31 December 2020 |
||||||||||||
|
Medicinal |
Psychedelic |
Unallocated |
Total |
|||||||||
|
Sales revenue |
1,177,163 |
|
— |
|
— |
|
1,177,163 |
|
||||
|
Product costs |
(537,939 |
) |
— |
|
— |
|
(537,939 |
) |
||||
|
Other income |
6 |
|
— |
|
52,072 |
|
52,078 |
|
||||
|
Expenses |
(2,157,611 |
) |
(90,000 |
) |
(1,333,080 |
) |
(3,580,691 |
) |
||||
|
Loss before tax |
(1,518,381 |
) |
(90,000 |
) |
(1,281,008 |
) |
(2,889,389 |
) |
||||
|
Segment assets |
774,229 |
|
— |
|
11,406,723 |
|
12,180,952 |
|
||||
|
Segment liabilities |
(119,141 |
) |
— |
|
(259,308 |
) |
(378,449 |
) |
||||
F-42
Notes to the Condensed Consolidated Financial Statements
for the Half-Years ended 31 December 2020 and 2019
NOTE 4: SEGMENT REPORTING (cont.)
|
6 months ended 31 December 2019 |
||||||||||||
|
Medicinal |
Dental |
Unallocated |
Total |
|||||||||
|
Sales revenue |
7,350 |
|
352,150 |
|
— |
|
359,500 |
|
||||
|
Product costs |
(8,450 |
) |
(204,334 |
) |
— |
|
(212,784 |
) |
||||
|
Other income |
— |
|
140,817 |
|
2,929 |
|
143,746 |
|
||||
|
Expenses |
(322,779 |
) |
(575,164 |
) |
(1,604,523 |
) |
(2,502,466 |
) |
||||
|
Loss before tax |
(323,879 |
) |
(286,531 |
) |
(1,601,594 |
) |
(2,212,004 |
) |
||||
|
Segment assets |
96,836 |
|
338,666 |
|
5,162,710 |
|
5,598,212 |
|
||||
|
Segment liabilities |
(130,330 |
) |
(241,441 |
) |
(17,768 |
) |
(389,539 |
) |
||||
____________
(1) Commenced 20 November 2020
(2) Ceased 30 June 2020
NOTE 5: SHARE BASED PAYMENTS
A. Securities on Issue at 30 June 2020
As at 30 June 2020, the Group had a number of securities on issue that had either not completed all of their vesting conditions or had not yet reached their performance hurdles (or both). These included:
a. 88,000,000 unlisted options previously issued with various performance hurdles set for achievement prior to their expiry date of 1 December 2020 did not meet these hurdles and were lapsed. The amount of $72,656 that had been expensed during FY20 has been written-back in the current period;
b. 1,166,666 ordinary shares approved by shareholders on 26 June 2020. Half of these vest upon continuing employment with the Company by the CEO on 30 June 2021 and the other half on 30 June 2022. $456 was expensed for these options during FY20, with $20,810 expensed during this period. $20,810 will be expensed in the second half of FY21, and $13,924 will be expensed in FY22;
c. 1,500,000 options with a strike price of $0.05 (750,000 expiring on 30 June 2025 and 750,000 expiring on 30 June 2026) were issued after approval by shareholders on 26 June 2020. Each 750,000 of these vest upon continued employment with the Company by the CEO until 30 June 2021 and 30 June 2022 respectively. $438 was expensed for these options during FY20, with $19,969 expensed during this period. $19,969 will be expensed in the second half of FY21, and $13,801 will be expensed in FY22;
d. 18,266,328 value-based performance rights with an expiry date of 22 November 2021, achieved their value milestones (ranging between $60m and $150m market capitalisation) and converted to ordinary shares during the period. Up to 30 June 2020, $127,235 had been expensed and a residual amount of $190,059 was to be expensed across the remainder of their vesting period, however having now vested, the full expense value of $190,059 has been recognised in the current period;
e. As at 31 December 2020, 12,037,265 value-based performance rights are yet to achieve their value milestone and need to do so prior to 22 November 2021 to convert to ordinary shares. At the start of the period, the amount of $60,964 had been expensed during FY20, and a value of $91,066 was yet to be expensed across the remainder of their vesting period (to 22 November 2021). Of this, $32,535 has been expensed in the current period;
F-43
Notes to the Condensed Consolidated Financial Statements
for the Half-Years ended 31 December 2020 and 2019
NOTE 5: SHARE BASED PAYMENTS (cont.)
f. 2,000,000 milestone-based performance rights subject to performance hurdles that must be achieved between 30 January 2021 and 31 March 2021 to convert to ordinary shares. $1,345 was expensed in FY20 with $54,789 being expensed in the first half of FY21 and the remaining $7,870 to be expensed in the second half of FY21. In the event that the performance hurdles are not achieved, this amount will be written back in the second half of FY21; and
g. 200,000,000 unlisted options issued that vest upon achievement of share price of $0.20 and expire on 30 September 2021. As at 30 June 2020 the amount of $131,096 had been expensed during FY20 with $175,203 to be expensed over their remaining life — of this $69,989 has been expensed during this period, with $105,214 to be expensed between 1 January 2021 and 30 September 2021.
|
Being expensed: |
|||||||||
|
Description |
During |
This period |
Remainder of |
FY22 |
|||||
|
88m unlisted options |
— |
(72,656 |
) |
— |
— |
||||
|
1.167m CEO ordinary shares |
456 |
20,810 |
|
20,810 |
13,924 |
||||
|
1.5m CEO unlisted options |
437 |
19,969 |
|
19,969 |
13,801 |
||||
|
18.266m value-based performance rights |
127,235 |
190,059 |
|
— |
— |
||||
|
12.037m value-based performance rights |
60,495 |
32,535 |
|
32,535 |
25,997 |
||||
|
2m milestone-based performance rights |
1,341 |
54,789 |
|
7,870 |
— |
||||
|
200m unlisted options |
130,667 |
69,989 |
|
69,989 |
35,224 |
||||
|
Share Based Payments expense (A) |
315,495 |
|
|||||||
B. New Securities Issued During Period
During the period, the Group also issued the following securities that are subject to expense at the time of their issue and over the life of their vesting period:
a. 2,952,619 ordinary shares approved by shareholders at a general meeting held on 26 June 2020;
b. 2,250,000 unlisted options approved by shareholders at a general meeting held on 26 June 2020;
c. 30,164,690 unlisted options issued on 2 October 2020 as consideration for broker support of the exercise of the 262m listed IHLOB options series; and
d. 20,000,000 unlisted options issued on 20 November 2020 as consideration for investor relations and corporate advisory work contracted.
|
Type |
Quantity |
Exercise |
Grant |
Vest date/ |
Expense |
||||||
|
Ordinary shares |
984,207 |
|
n/a |
1-Jul-2020 |
30-Jun-2021 |
48,226 |
|||||
|
Ordinary shares |
984,206 |
|
n/a |
1-Jul-2020 |
30-Jun-2022 |
43,403 |
|||||
|
Ordinary shares |
984,206 |
|
n/a |
1-Jul-2020 |
30-Jun-2023 |
36,170 |
|||||
|
Total (a) |
2,952,619 |
|
127,799 |
||||||||
|
Unlisted options |
750,000 |
$ |
0.05 |
1-Jul-2020 |
30-Jun-2025 |
25,432 |
|||||
|
Unlisted options |
750,000 |
$ |
0.05 |
1-Jul-2020 |
30-Jun-2026 |
27,450 |
|||||
|
Unlisted options |
750,000 |
$ |
0.05 |
1-Jul-2020 |
30-Jun-2027 |
29,040 |
|||||
|
Total (b) |
2,250,000 |
|
81,922 |
||||||||
|
Unlisted options |
30,164,690 |
$ |
0.08 |
2-Oct-2020 |
30-Sep-2021 |
876,284 |
|||||
|
Total (c) |
30,164,690 |
|
876,284 |
||||||||
|
Unlisted options |
10,000,000 |
$ |
0.15 |
20-Nov-2020 |
20-Nov-2024 |
659,400 |
|||||
|
Unlisted options |
10,000,000 |
$ |
0.25 |
20-Nov-2020 |
20-Nov-2024 |
539,500 |
|||||
|
Total (d) |
20,000,000 |
|
1,198,900 |
||||||||
F-44
Notes to the Condensed Consolidated Financial Statements
for the Half-Years ended 31 December 2020 and 2019
NOTE 5: SHARE BASED PAYMENTS (cont.)
|
Type |
Quantity |
Expense |
This period |
Remainder |
FY22 and |
|||||
|
(a) Ordinary Shares |
2,952,619 |
127,799 |
40,650 |
40,650 |
45,609 |
|||||
|
(b) Unlisted options |
2,250,000 |
81,922 |
24,226 |
24,226 |
32,940 |
|||||
|
(c) Unlisted options |
30,164,690 |
876,284 |
217,261 |
436,935 |
222,089 |
|||||
|
(d) Unlisted options |
20,000,000 |
1,198,900 |
44,890 |
198,174 |
955,835 |
|||||
|
Share Based Payments expense (B) |
327,027 |
C. Amount expensed as Share Based Payments expense in the half-year ended 31 December 2020 (Unaudited)
|
Share Based Payments expense (A) – for securities on issue at 30 June 2020 |
315,495 |
|
|
|
Share Based Payments expense (B) – for securities issued during this period |
327,027 |
|
|
|
Less amount charged to share raising costs |
(217,261 |
) |
|
|
Less amount charged to advertising and investor relations |
(44,890 |
) |
|
|
Total Share Based Payments expense |
380,371 |
|
D. Valuation assumptions
OPTIONS
The fair value of the equity-settled share options granted in the above tables is estimated as at the date of grant using a trinomial option model taking into account the terms and conditions upon which the options were granted.
|
Expiry date |
30-Sep-21 |
20-Nov-23 |
20-Nov-23 |
30-Jun-25 |
30-Jun-26 |
30-Jun-27 |
||||||||||||||||||
|
Exercise price |
$ |
0.08 |
|
$ |
0.15 |
|
$ |
0.25 |
|
$ |
0.05 |
|
$ |
0.05 |
|
$ |
0.05 |
|
||||||
|
Dividend yield |
|
0 |
% |
|
0 |
% |
|
0 |
% |
|
0 |
% |
|
0 |
% |
|
0 |
% |
||||||
|
Expected volatility |
|
100 |
% |
|
100 |
% |
|
100 |
% |
|
100 |
% |
|
100 |
% |
|
100 |
% |
||||||
|
Risk-free interest rate |
|
2 |
% |
|
2 |
% |
|
2 |
% |
|
2 |
% |
|
2 |
% |
|
2 |
% |
||||||
|
Expected life of option |
|
1 |
|
|
3 |
|
|
3 |
|
|
4 |
|
|
5 |
|
|
6 |
|
||||||
|
Grant date share price |
$ |
0.077 |
|
$ |
0.115 |
|
$ |
0.115 |
|
$ |
0.049 |
|
$ |
0.049 |
|
$ |
0.049 |
|
||||||
The expected life of the options is based on historical data and is not necessarily indicative of exercise patterns that may occur. The expected volatility reflects the assumption that the historical volatility is indicative of future trends, which may also not necessarily be the actual outcome. The options vest upon the shares having a closing price of 20 cents per share or more for any 5 trading days at any time from the date of grant of the options until the expiry date of the options (30 September 2021).
SHARES
Ordinary shares issued have been valued based on the market price of the shares on grant date.
NOTE 6: DIVIDENDS
No dividend have been declared or paid in the half-years ended 31 December 2020 or 2019.
F-45
Notes to the Condensed Consolidated Financial Statements
for the Half-Years ended 31 December 2020 and 2019
NOTE 7: ISSUED CAPITAL (UNAUDITED)
|
Movement in: |
Issued |
Number of securities: |
||||||||||||||
|
Ordinary |
Performance |
Performance |
Listed |
Unlisted |
||||||||||||
|
As at 1 July 2019 |
26,951,744 |
|
581,897,040 |
20,000,002 |
24,166,668 |
|
262,960,728 |
|
— |
|
||||||
|
Placement shares issued |
6,885,354 |
|
114,663,460 |
— |
— |
|
— |
|
— |
|
||||||
|
Shares issued on exercise of options |
234,217 |
|
10,855,423 |
— |
— |
|
(855,423 |
) |
(10,000,000 |
) |
||||||
|
Share issue costs |
(493,385 |
) |
— |
— |
— |
|
— |
|
— |
|
||||||
|
Options granted |
— |
|
— |
— |
— |
|
— |
|
209,919,705 |
|
||||||
|
As at 31 December 2019 |
33,577,930 |
|
707,415,923 |
20,000,002 |
24,166,668 |
|
262,105,305 |
|
199,919,705 |
|
||||||
|
|
|
|
|
|||||||||||||
|
As at 1 July 2020 |
34,192,043 |
|
748,654,489 |
— |
41,553,593 |
|
260,533,407 |
|
380,169,705 |
|
||||||
|
Shares issued on exercise of options |
11,199,678 |
|
270,262,674 |
— |
— |
|
(260,533,407 |
) |
(9,729,267 |
) |
||||||
|
Performance rights converted |
— |
|
18,266,328 |
— |
(18,266,328 |
) |
— |
|
— |
|
||||||
|
Share issue costs |
(315,237 |
) |
— |
— |
— |
|
— |
|
— |
|
||||||
|
Options granted |
— |
|
— |
— |
— |
|
— |
|
52,414,690 |
|
||||||
|
Other securities issued |
— |
|
2,952,619 |
— |
— |
|
— |
|
— |
|
||||||
|
Lapsed/expired |
— |
|
— |
— |
— |
|
— |
|
(88,000,000 |
) |
||||||
|
As at 31 December 2020 |
45,076,484 |
|
1,040,136,110 |
— |
23,287,265 |
|
— |
|
334,855,128 |
|
||||||
|
Expiry date and exercise price of options |
As at |
As at |
As at |
As at |
||||
|
01-May-2020 $0.03 unlisted |
— |
10,000,000 |
— |
— |
||||
|
01-Dec-2020 $0.04 unlisted |
— |
12,000,000 |
— |
— |
||||
|
30-Sep-2020 $0.04 IHLOB |
262,105,305 |
— |
— |
— |
||||
|
30-Jun-2025 $0.05 unlisted |
— |
— |
— |
1,500,000.00 |
||||
|
30-Jun-2026 $0.05 unlisted |
— |
— |
— |
1,500,000.00 |
||||
|
30-Jun-2027 $0.05 unlisted |
— |
— |
— |
1,500,000.00 |
||||
|
01-Dec-2020 $0.06 unlisted |
— |
14,000,000 |
— |
— |
||||
|
01-Dec-2020 $0.08 unlisted |
— |
16,000,000 |
— |
— |
||||
|
30-Sep-2021 $0.08 unlisted |
— |
89,919,705 |
— |
110,355,128 |
||||
|
01-Dec-2020 $0.10 unlisted |
— |
18,000,000 |
— |
— |
||||
|
01-Dec-2020 $0.12 unlisted |
— |
20,000,000 |
— |
— |
||||
|
01-Dec-2020 $0.14 unlisted |
— |
20,000,000 |
— |
— |
||||
|
23-Nov-2023 $0.15 unlisted |
— |
— |
— |
10,000,000 |
||||
|
23-Nov-2023 $0.25 unlisted |
— |
— |
— |
10,000,000 |
||||
|
30-Sep-2021 $0.20 unlisted |
— |
— |
— |
200,000,000 |
||||
|
Total |
262,105,305 |
199,919,705 |
— |
334,855,128 |
NOTE 8: CONTINGENCIES
There has been no change in contingent liabilities since the last annual reporting date.
F-46
Notes to the Condensed Consolidated Financial Statements
for the Half-Years ended 31 December 2020 and 2019
NOTE 9: FINANCIAL INSTRUMENTS
The Group has a number of financial instruments which are not measured at fair value in the condensed consolidated statement of financial position.
The Directors consider that the carrying amounts of current receivables, current payables and current borrowings are considered to be a reasonable approximation of their fair values.
NOTE 10: RELATED PARTY DISCLOSURES
Directors’ holdings in securities
|
31 December 2020 (Unaudited) |
Options |
Performance |
Ordinary |
|||
|
Mr Troy Valentine |
7,116,950 |
1,500,000 |
23,734,248 |
|||
|
Mr Peter Widdows |
657,895 |
1,500,000 |
15,915,799 |
|||
|
Dr Sud Agarwal* |
200,000,000 |
14,037,265 |
54,266,328 |
|||
|
Mr Joel Latham |
4,700,000 |
5,000,000 |
17,948,414 |
|
31 December 2019 (Unaudited) |
Options |
Performance |
Ordinary |
|||
|
Mr Troy Valentine |
48,355,557 |
2,762,538 |
19,900,914 |
|||
|
Mr Peter Widdows |
3,957,895 |
1,833,334 |
12,282,456 |
|||
|
Dr Sud Agarwal* |
110,000,000 |
— |
10,000,000 |
|||
|
Mr Joel Latham |
4,437,500 |
6,000,000 |
10,245,795 |
____________
* Options and shares reported for Dr Sud Agarwal include those owned by Cannvalate Pty Ltd — an entity of which Dr Agarwal is a significant shareholder, the CEO and a director.
# Performance Shares convert on one-for-one basis on achievement of sales targets — refer to 30 June 2020 financial statements for further details. Performance Rights convert on a one-for one basis on achievement of sales targets or EBITDA hurdles — refer to 30 June 2020 financial statements for further details.
Other Related Party Disclosures
There were no other transactions during the half-years with related parties and there were no liabilities due to related parties at the end of the half-years.
NOTE 11: SIGNIFICANT EVENTS AFTER BALANCE DATE
There has not been any other matter or circumstance that has arisen after balance date that has significantly affected, or may significantly affect, the operations of the Group, the results of those operations, or the state of affairs of the Group in future financial periods.
NOTE 12: DISCONTINUED OPERATIONS
As disclosed in the Group’s financial report for the year ended 30 June 2020, the Group sold its 100% subsidiary, Gameday International Pty Ltd (“Gameday”), on 30 June 2020. The Condensed Consolidated Statement of Comprehensive Income discloses a loss after tax from discontinued operations, being Gameday, of $286,531 for the half year ended 31 December 2019. This amount represents the net loss attributed to Gameday for that period.
F-47
American Depositary Shares
Representing Ordinary Shares
_____________________________________
PRELIMINARY PROSPECTUS
, 2021
_____________________________________
Roth Capital Partners, LLC
Through and including , 2021 (the 25th day after the date of this prospectus), all dealers effecting transactions in the ADSs, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to a dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to an unsold allotment or subscription.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 6. Indemnification of Directors and Officers.
Australian law. Australian law provides that a company or a related body corporate of the company may provide for indemnification of officers and directors, except to the extent of any of the following liabilities incurred as an officer or director of the company:
• a liability owed to the company or a related body corporate of the company;
• a liability for a pecuniary penalty order made under section 1317G or a compensation order under section 961M, 1317H, 1317HA, 1317HB 1317HC or 1317HE of the Corporations Act;
• a liability that is owed to someone other than the company or a related body corporate of the company and did not arise out of conduct in good faith; or
• legal costs incurred in defending an action for a liability incurred as an officer or auditor of the company if the costs are incurred:
• in defending or resisting proceedings in which the person is found to have a liability for which they cannot be indemnified as set out above;
• in defending or resisting criminal proceedings in which the person is found guilty;
• in defending or resisting proceedings brought by the Australian Securities& Investments Commission or a liquidator for a court order if the grounds for making the order are found by the court to have been established (except costs incurred in responding to actions taken by the Australian Securities& Investments Commission or a liquidator as part of an investigation before commencing proceedings for a court order); or
• in connection with proceedings for relief to the person under the Corporations Act 2001 (Cth), or the Corporations Act, in which the court denies the relief.
Constitution. Our Constitution provides that, except to the extent prohibited by the law and the Corporations Act and, to the extent that the officer is not otherwise indemnified by us pursuant to an indemnity, we indemnify every person who is or has been an officer of the company against any liability or claim (other than legal costs that are unreasonable) incurred by that person as an officer. This includes any liability or claim incurred by that person in their capacity as an officer of a subsidiary of the company where the company requested that person to accept that appointment.
SEC Position. Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
ITEM 7. Recent Sales of Unregistered Securities.
Since July 1, 2018, the following changes have been made to our ordinary share capital:
• the Registrant granted share options to purchase an aggregate of 968,279,897 ordinary shares with a weighted-average exercise price of US$ 0.0654 per share to employees, directors, officers and consultants, under Regulation S. Options to purchase an aggregate of 53,685,260 ordinary have been exercised for aggregate consideration of approximately US$ 1,865,307, under Regulation S;
II-1
• the Registrant granted performance rights equivalent to an aggregate of 81,303,593 ordinary shares to employees, directors, officers and consultants, under Regulation S. Performance rights equivalent to 36,250,001 ordinary shares have been exercised for no consideration, under Regulation S;
• on August 29, 2018, the Registrant issued 6,000,000 ordinary shares at a price of A$0.025 per share to institutional investors, under Regulation S;
• on September 20, 2018, the Registrant issued 16,800,000 ordinary shares at a price of A$0.20 per share to institutional investors, under Regulation S;
• on October 22, 2018, the Registrant issued 33,117,189 ordinary shares at a price of A$0.01 per share to institutional investors, under Regulation S;
• on November 7, 2018, the Registrant issued 40,454,873 ordinary shares at a price of A$0.01 per share to institutional investors, under Regulation S;
• on November 21, 2018, the Registrant issued 85,534,312 ordinary shares at a price of A$0.01 per share to institutional investors, under Regulation S;
• on January 31, 2019, the Registrant issued 74,100,000 ordinary shares at a price of A$0.01 per share and 3,500,000 ordinary shares at a price of A$0.02 to institutional investors, under Regulation S;
• on April 26, 2019, the Registrant issued 9,269,086 ordinary shares at a price of A$0.01 per share to institutional investors, under Regulation S;
• on July 8, 2019, the Registrant issued 31,983,470 ordinary shares and 2,000,000 ordinary shares, at a price of A$0.038 per share and A$0.02 to institutional investors, under Regulation S;
• on October 25, 2019, the Registrant issued 64,103,564 ordinary shares at a price of A$0.078 per share to institutional investors, under Regulation S;
• on December 31, 2019, the Registrant issued 10,000,000 ordinary shares at a price of A$0.04 per share to institutional investors, under Regulation S;
• on June 29, 2020, the Registrant issued 1,750,000 ordinary shares at a price of A$0.016 per share to institutional investors, under Regulation S;
• on June 29, 2020, the Registrant issued 4,000,000 ordinary shares at a price of A$0.048 per share to institutional investors, under Regulation S;
• on July 1, 2020, the Registrant issued 2,952,619 ordinary shares at a price of A$0.036 per share to institutional investors, under Regulation S;
None of the foregoing transactions involved any underwriter, underwriting discounts or commissions, or any public offering. The recipients of the securities in each of these transactions represented their intentions to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof, and appropriate legends were placed on the share certificates issued in these transactions. All recipients had adequate access, through their relationships with us, to information about us. The sales of these securities were made without any general solicitation or advertising.
II-2
ITEM 8. Exhibits and Financial Statement Schedules.
(a) Exhibits
|
Exhibit |
Description |
|
|
1.1* |
Form of Underwriting Agreement |
|
|
3.1# |
||
|
4.1* |
Form of Deposit Agreement between Incannex Healthcare Limited and Deutsche Bank Trust Company Americas as Depositary |
|
|
4.2* |
Form of American Depositary Receipt (included in Exhibit 4.1) |
|
|
4.3* |
Form of Underwriter’s Warrant |
|
|
5.1* |
Opinion of Rimôn Law |
|
|
10.1# |
Employment Agreement between Incannex Healthcare Limited and Joel Latham, dated July 1, 2020 |
|
|
10.2# |
||
|
10.3# |
Service Agreement between Incannex Healthcare Limited and Madhukar Bhalla, dated June 28, 2021 |
|
|
10.4✓ |
||
|
10.5✓ |
||
|
10.6✓ |
||
|
10.7✓ |
||
|
10.8✓ |
||
|
10.9✓ |
||
|
10.10✓ |
||
|
10.11✓ |
||
|
21.1# |
||
|
23.1 |
||
|
23.2* |
Consent of Rimôn Law (included in Exhibit 5.1) |
|
|
24.1# |
Power of Attorney (included in signature page to Registration Statement) |
____________
* to be filed by amendment
# previously filed
✓ Certain confidential information in this exhibit was omitted by means of marking such information with brackets (“[***]”) because the identified confidential information is not material and is the type that the registrant treats as private or confidential.
ITEM 9. Undertakings.
The undersigned Registrant hereby undertakes to provide to the underwriter at the closing specified in the underwriting agreement, certificates in such denominations and registered in such names as required by the underwriter to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such
II-3
director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned Registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this Registration Statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this Registration Statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-4
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form F-1 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized in Docklands, Australia on September 28, 2021.
|
Incannex Healthcare Limited |
||||
|
By: |
/s/ Joel Latham |
|||
|
Name: |
Joel Latham |
|||
|
Title: |
Chief Executive Officer and Managing Director |
|||
Pursuant to the requirements of the Securities Act of 1933, as amended, this Amendment No. 1 to the registration statement has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
|
Signature |
Title |
Date |
||
|
/s/ Joel Latham |
Chief Executive Officer and Managing Director |
September 28, 2021 |
||
|
Joel Latham |
(Principal Executive Officer) |
|||
|
* |
Chief Financial Officer and Company Secretary |
September 28, 2021 |
||
|
Madhukar Bhalla |
(Principal Financial and Accounting Officer) |
|||
|
* |
Director |
September 28, 2021 |
||
|
Troy Valentine |
||||
|
* |
Director |
September 28, 2021 |
||
|
Dr. Sud Agarwal |
||||
|
* |
Director |
September 28, 2021 |
||
|
Peter Widdows |
|
*By: |
/s/ Joel Latham |
|||
|
Joel Latham |
II-5
Signature of Authorized U.S. Representative of the Registrant
Pursuant to the requirements of the Securities Act of 1933, as amended, the undersigned, the duly authorized representative in the United States of Incannex Healthcare Limited, has signed this Amendment No. 1 to the Registration Statement on September 28, 2021.
|
By: |
/s/ Donald J. Puglisi |
|||
|
Name: |
Donald J. Puglisi |
|||
|
Title: |
Managing Director |
II-6