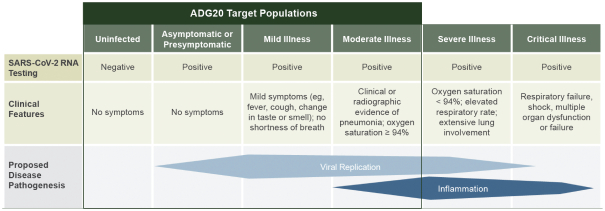
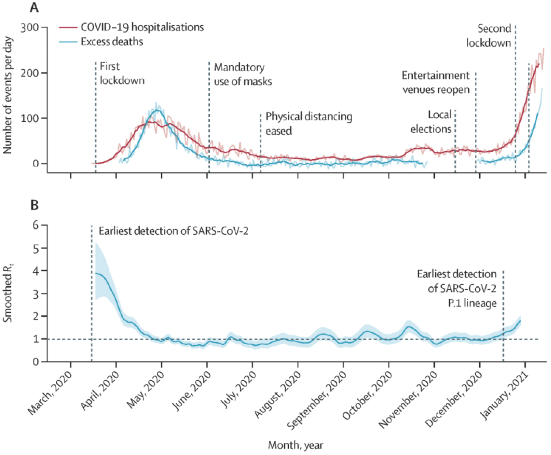
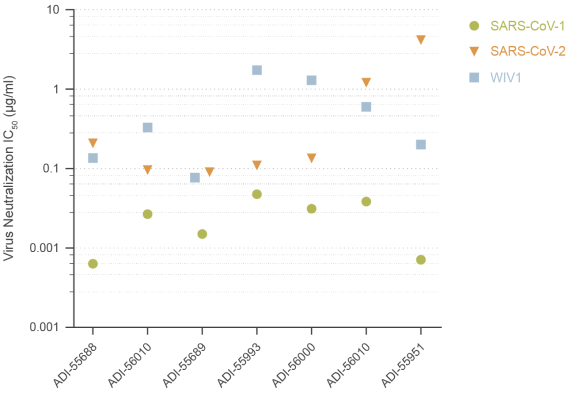
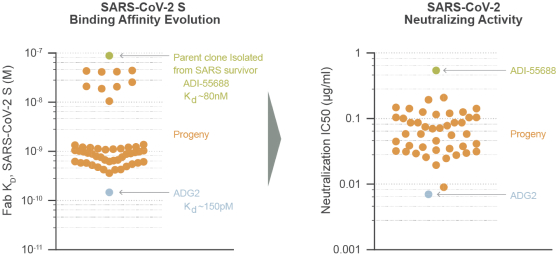
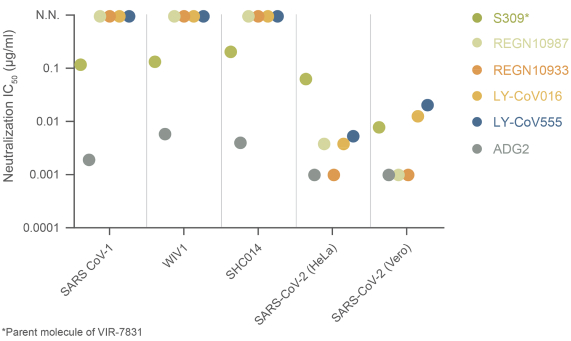
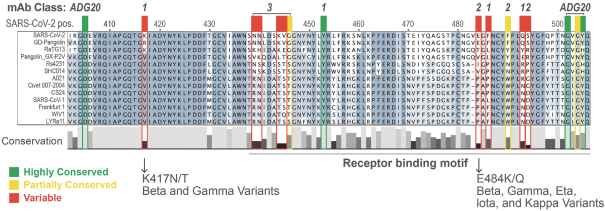
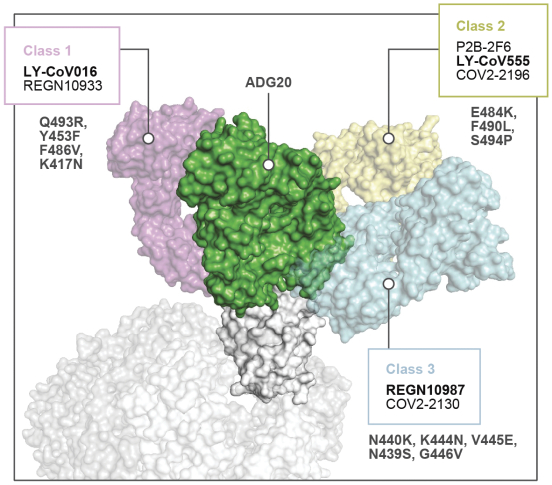
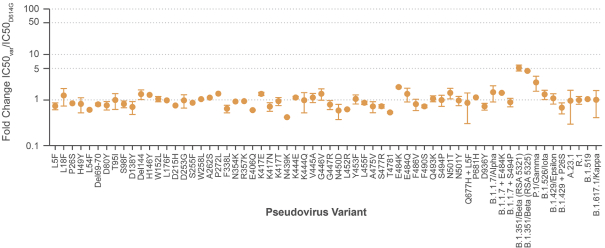
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION
DATED AUGUST 2, 2021
17,700,000 Shares
Adagio Therapeutics, Inc.
COMMON STOCK
Adagio Therapeutics, Inc. is offering 17,700,000 shares of common stock. This is our initial public offering and no public market exists for our common stock. We anticipate that the initial public offering price will be between $16.00 and $18.00 per share.
We have applied to list our common stock on The Nasdaq Global Market under the trading symbol “ADGI.”
We are an “emerging growth company” and a “smaller reporting company” as defined under U.S. federal securities laws and, as such, will be subject to reduced public company reporting requirements for this prospectus and future filings. See “Prospectus Summary—Implications of Being an Emerging Growth Company and a Smaller Reporting Company.”
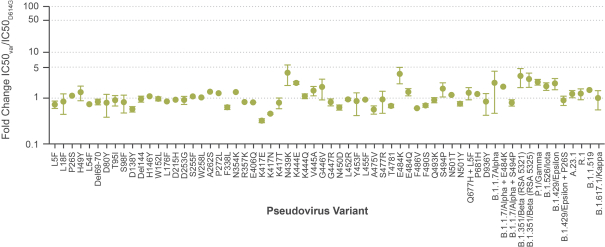
Investing in our common stock involves risks. See “Risk Factors” beginning on page 13 of this prospectus.
|
Per Share |
Total |
|||||||
| Initial Public Offering Price |
$ | $ | ||||||
| Underwriting Discounts and Commissions (1) |
$ | $ | ||||||
| Proceeds, before expenses, to us |
$ | $ | ||||||
| (1) | We refer you to “Underwriting” for additional information regarding total underwriter compensation. |
We have granted the underwriters an option for a period of 30 days to purchase up to an additional 2,655,000 shares of our common stock.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The underwriters expect to deliver the shares of common stock against payment in New York, New York on or about , 2021.
Joint Book-Running Managers
| MORGAN STANLEY | JEFFERIES | STIFEL | GUGGENHEIM SECURITIES |
The date of this prospectus is , 2021