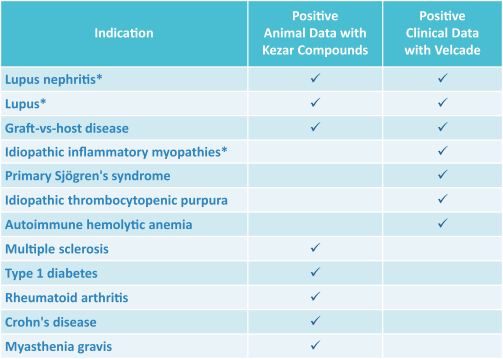
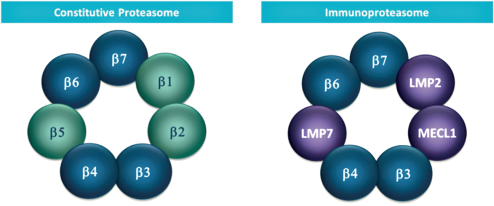
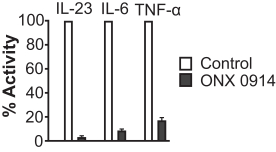
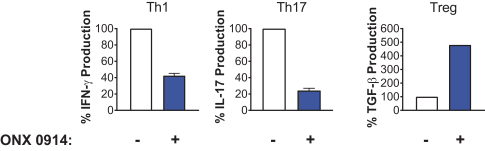
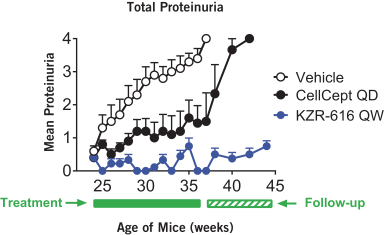
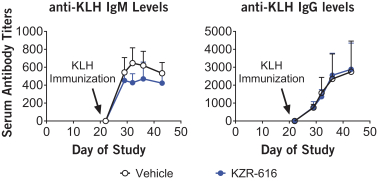
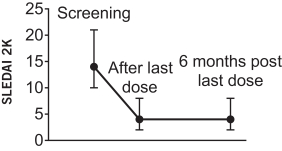
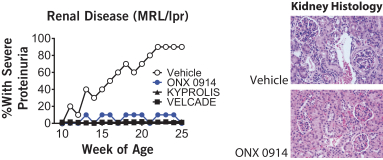
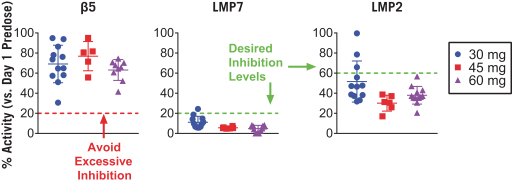
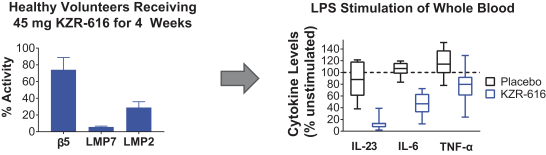
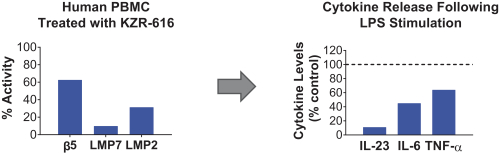
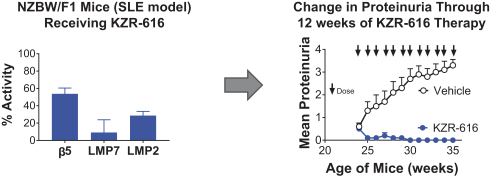
additional product candidates we may pursue. Our expenses could increase beyond expectations if the U.S. Food and Drug Administration, or FDA, or other regulatory authorities require us to
perform clinical and other studies in addition to those that we currently anticipate. In addition, if we obtain marketing approval for our product candidates, we expect to incur significant expenses related to sales, marketing, manufacturing and
distribution. Furthermore, upon the completion of this offering, we expect to incur additional costs associated with operating as a public company.
As of
March 31, 2018, our cash and cash equivalents was $47.1 million. We believe that the net proceeds from this offering, together with our existing cash and cash equivalents, will fund our current operating plans through at least the next 12
months from the date the financial statements were issued. However, our operating plan may change as a result of many factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or
debt financings, third-party funding, marketing and distribution arrangements, as well as other collaborations, strategic alliances and licensing arrangements, or any combination of these approaches.
In any event, we will require substantial additional capital to develop a delivery system for KZR-616, conduct additional
clinical trials, seek regulatory approval and commence commercialization of KZR-616 or any future product candidates. Even if we believe we have sufficient capital for our current or future operating plans, we
may seek additional capital if market conditions are favorable or if we have specific strategic considerations. Any additional capital raising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize KZR-616 and any future product candidates.
If we do not raise additional capital in sufficient amounts, or on terms acceptable to us, we may be prevented from pursuing discovery, development and
commercialization efforts, which will harm our business, operating results and prospects.
Raising additional capital may cause dilution to our stockholders,
including purchasers of shares of our common stock in this offering, restrict our operations or require us to relinquish proprietary rights.
Until such
time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through public or private equity or debt financings, third-party funding, marketing and distribution arrangements, as well as other collaborations,
strategic alliances and licensing arrangements, or any combination of these approaches. We do not have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities,
your ownership interest may be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a stockholder. In addition, we may issue equity or debt securities as consideration for
obtaining rights to additional compounds.
Debt and equity financings, if available, may involve agreements that include covenants limiting or restricting our
ability to take specific actions, such as redeeming our shares, making investments, incurring additional debt, making capital expenditures, declaring dividends or placing limitations on our ability to acquire, sell or license intellectual property
rights and other operating restrictions that could negatively impact our ability to conduct our business. If we raise additional capital through future collaborations, strategic alliances or third-party licensing arrangements, we may have to
relinquish valuable rights to our intellectual property, future revenue streams, research programs or product candidates, or grant licenses on terms that may not be favorable to us.
If we are unable to raise additional capital when needed, we may be required to delay, limit, reduce or terminate our drug development or future commercialization
efforts, or grant rights to develop and market product candidates that we would otherwise develop and market ourselves.
We may be required to make
significant payments in connection with our license agreement with Onyx Therapeutics, Inc., or Onyx, for KZR-616 and other compounds.
In June 2015, we acquired rights to KZR-616, pursuant to a license agreement with Onyx, or the Onyx license agreement. Under the
Onyx license agreement, we are subject to significant obligations, including payment obligations triggered upon achievement of specified milestones and royalties on licensed product sales, as well as other material obligations. We are obligated to
pay Onyx milestone payments up to an aggregate of $172.5 million upon the achievement of certain development, regulatory and sales milestone events. In addition, we are obligated to pay Onyx tiered royalties based on net sales of KRZ-616. If these payments become due, we may not have sufficient funds available to meet our obligations and our development efforts may be harmed.
13