
As filed with the Securities and Exchange Commission December 1, 2016
Registration Statement No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER THE SECURITIES ACT OF 1933
RECRO PHARMA, INC.
(Exact name of registrant as specified in its charter)
| Pennsylvania | 2834 | 26-1523233 | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification No.) |
490 Lapp Road
Malvern, PA 19355
(484) 395-2400
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Gerri A. Henwood
President and Chief Executive Officer
Recro Pharma, Inc.
490 Lapp Rd
Malvern, PA 19355
(484) 395-2400
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
| Rachael M. Bushey, Esq. Hogan Lovells US LLP 1835 Market Street, 29th Floor Philadelphia, PA 19103 (267) 675-4675 |
David S. Rosenthal, Esq. Dechert LLP 1095 Avenue of the Americas New York, New York 10036 (212) 698-3500 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after this registration statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer ¨ | Non-accelerated filer ¨ (Do not check if a smaller reporting company) |
Smaller Reporting Company þ |
The registrant is an emerging growth company, as defined in Section 2(a) of the Securities Act. This Registration Statement complies with the requirements that apply to an issuer that is an emerging growth company.
|
| ||||
| Title of each class of securities to be registered |
Proposed Maximum Aggregate Offering Price(1) |
Amount of Registration Fee(2) | ||
| Common Stock, par value $0.01 per share |
$57,500,000 | $6,664.25 | ||
|
| ||||
|
| ||||
| (1) | Estimated solely for purposes of computing the amount of the registration fee pursuant to Rule 457(o) under the Securities Act of 1933, as amended. Includes shares subject to the underwriters’ option to purchase additional shares. |
| (2) | Calculated pursuant to Rule 457(o) based on an estimate of the proposed maximum aggregate offering price of the securities registered hereunder. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
The information contained in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject To Completion, Dated December 1, 2016
PRELIMINARY PROSPECTUS
Shares
RECRO PHARMA, INC.
Common Stock
$ per share
| • Recro Pharma, Inc. is offering shares of common stock.
• The last reported sale price for our common stock on the Nasdaq Capital Market on November 30, 2016 was $8.00 per share. |
• Trading Symbol: Nasdaq Capital Market—REPH. |
We are an “emerging growth company” as defined by the Jumpstart Our Business Startups Act of 2012 and, as such, we are eligible for reduced public company reporting requirements. Please see “Summary—Implications of Being an Emerging Growth Company.”
Investing in our common stock involves a high degree of risk. See “Risk Factors” beginning on page 10 of this prospectus and under similar headings in the documents incorporated by reference into this prospectus.
| Per Share | Total | |||||||
| Public offering price |
$ | $ | ||||||
| Underwriting discounts and commissions(1) |
$ | $ | ||||||
| Proceeds, before expenses, to us |
$ | $ | ||||||
| (1) | See “Underwriting” for additional disclosure regarding underwriting discounts, commissions and estimated offering expenses, including expenses for which we have agreed to reimburse the underwriters. |
We have granted the underwriters an option for a period of 30 days to purchase an additional shares of our common stock. If the underwriters exercise the option in full, the total underwriting discounts and commissions payable by us will be $ , and the total proceeds to us, before expenses, will be $ .
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Delivery of the shares of common stock is expected to be made on or about , 2016.
Piper Jaffray
The date of this prospectus is , 2016.
| Page | ||||
| 1 | ||||
| 10 | ||||
| 12 | ||||
| 13 | ||||
| 14 | ||||
| 15 | ||||
| 16 | ||||
| 46 | ||||
| 48 | ||||
| 50 | ||||
| 52 | ||||
| 54 | ||||
| 58 | ||||
| 66 | ||||
| 66 | ||||
| 67 | ||||
| 68 | ||||
You should read this prospectus, including the information incorporated by reference herein, and any related free writing prospectus that we have authorized for use in connection with this offering.
You should rely only on the information that we have included or incorporated by reference in this prospectus and any related free writing prospectus that we may authorize to be provided to you. We have not authorized any dealer, salesman or other person to give any information or to make any representation other than those contained or incorporated by reference in this prospectus or any related free writing prospectus that we may authorize to be provided to you. You must not rely upon any information or representation not contained or incorporated by reference in this prospectus or any related free writing prospectus. This prospectus and any related free writing prospectus do not constitute an offer to sell or the solicitation of an offer to buy any securities other than the registered securities to which they relate, nor do this prospectus or any related free writing prospectus constitute an offer to sell or the solicitation of an offer to buy securities in any jurisdiction to any person to whom it is unlawful to make such offer or solicitation in such jurisdiction.
You should not assume that the information contained in this prospectus or any related free writing prospectus is accurate on any date subsequent to the date set forth on the front of the document or that any information we have incorporated by reference herein or therein is correct on any date subsequent to the date of the document incorporated by reference, even though this prospectus or any related free writing prospectus is delivered, or securities are sold, on a later date.
This prospectus contains or incorporates by reference summaries of certain provisions contained in some of the documents described herein, but reference is made to the actual documents for complete information. All of the summaries are qualified in their entirety by the actual documents. Copies of some of the documents referred to herein have been filed or have been incorporated by reference as exhibits to the registration statement of which this prospectus forms a part, and you may obtain copies of those documents as described in this prospectus under the heading “Where You Can Find More Information.”
This summary highlights information contained in other parts of this prospectus and in the documents we incorporate by reference. Because it is only a summary, it does not contain all of the information that you should consider before investing in shares of our common stock and it is qualified in its entirety by, and should be read in conjunction with, the more detailed information appearing elsewhere in this prospectus, any applicable free writing prospectus and the documents incorporated by reference herein and therein. You should read all such documents carefully, especially the risk factors and our consolidated financial statements and the related notes included or incorporated by reference herein or therein, before deciding to buy shares of our common stock. Unless the context requires otherwise, references in this prospectus to “Recro,” “we,” “us” and “our” refer to Recro Pharma, Inc. and our subsidiaries.
Overview
Company Overview
We are a revenue-generating, specialty pharmaceutical company primarily focused on developing innovative products for hospitals and ambulatory care settings. Our lead product candidate, injectable meloxicam, is a proprietary injectable form of meloxicam, a long-acting preferential COX-2 inhibitor that has successfully completed five clinical trials in the treatment of moderate to severe pain, including a two recently completed pivotal Phase III clinical trials. As injectable meloxicam is not in the opioid class of drugs, we believe it will overcome many of the issues associated with commonly prescribed opioid therapeutics, including addiction, misuse/diversion, respiratory distress and constipation while maintaining analgesic, or pain relieving, effect.
In addition to developing proprietary drug candidates, we, through our subsidiary, Recro Gainesville LLC, or Recro Gainesville, leverage our formulation expertise to develop and manufacture pharmaceutical products using our proprietary delivery technologies for commercial partners who commercialize or plan to commercialize these products. These collaborations result in revenue streams including royalties, profit sharing, research and development and manufacturing, which support continued operations for Recro Gainesville as well as our research and development of proprietary product candidates.
Recent Developments
In November 2016, we announced positive results from the second of our two pivotal Phase III clinical trials for intravenous, or IV, meloxicam, evaluating pain relief over a 24-hour period in a soft tissue, post-operative pain model (abdominoplasty). In the trial, IV meloxicam achieved the primary endpoint of a statistically significant difference in Summed Pain Intensity Difference, or SPID, over the first 24 hours, or SPID24, compared to placebo. In this multicenter, randomized, double-blind, placebo-controlled clinical trial, 219 patients were enrolled and randomly assigned to receive a postoperative regimen of IV meloxicam (30mg bolus injection) or placebo in a 1:1 ratio, once every 24 hours. The IV meloxicam treatment arm demonstrated a statistically significant reduction in SPID24 (p=0.0145) compared to the placebo arm. With the positive data from this study, we believe this completes the efficacy program for the IV meloxicam new drug application, or NDA.
Lead Product Candidate – Injectable Meloxicam
Meloxicam is a long-acting, preferential COX-2 inhibitor that possesses anti-inflammatory, analgesic, and antipyretic activities, which are believed to be related to the inhibition of cyclooxygenase, or COX,
1
and subsequent reduction in prostaglandin biosynthesis. Meloxicam has been marketed by Boehringer Ingelheim Pharmaceuticals, Inc. since the 1990s as an oral agent, Mobic®. Mobic tablets and suspension are indicated for the relief of the signs and symptoms of osteoarthritis and rheumatoid arthritis and the relief of the signs and symptoms of pauciarticular or polyarticular juvenile rheumatoid arthritis in patients 2 years or older. We believe that IV meloxicam compares favorably to competitive therapies in onset of pain relief, duration of pain relief, extent of pain relief and time to peak analgesic effect.
In early 2016, based on feedback from the U.S. Food and Drug Administration, or FDA, we commenced our Phase III clinical trial program for IV meloxicam. The program includes two pivotal Phase III clinical trials, both of which IV meloxicam has successfully completed. In July 2016, we announced positive results from one pivotal clinical trial, evaluating pain relief over a 48-hour period in a hard tissue, post-operative pain model (bunionectomy). In the trial, IV meloxicam achieved the primary endpoint of a statistically significant difference in SPID over the first 48 hours, or SPID48, compared to placebo. In this multicenter, randomized, double-blind, placebo-controlled clinical trial, 201 patients were enrolled and randomly assigned to receive a postoperative regimen of IV meloxicam (30mg bolus injection over 15-30 seconds) or placebo in a 1:1 ratio, once every 24 hours for up to three doses following bunionectomy surgery, a representative hard tissue surgery. The IV meloxicam treatment arm demonstrated a statistically significant reduction in SPID48 (p=0.0034) compared to the placebo arm. The study also achieved 15 of the 19 secondary endpoints, including statistically significant differences in SPID6 (p=0.0153), SPID12 (p=0.0053), SPID24 (p=0.0084), SPID24-48 (p=0.0050), time to first use of rescue medication (p=0.0076), and several other rescue use and pain relief metrics during the first 48 hours, compared to placebo. The safety results demonstrated that IV meloxicam was well tolerated with no serious adverse events, or SAEs, or bleeding events in the IV meloxicam-treated patients. The most common adverse events, or AEs, occurring in at least 3% of IV meloxicam-treated treated patients, were nausea, headache, pruritus, constipation vomiting, dizziness, flushing and somnolence, and were comparable to the placebo group. The IV meloxicam-treated patients experienced injection site pain and injection site erythema at a rate comparable to placebo. The majority of treatment emergent AEs, or TEAEs, were mild in nature and there were no discontinuations due to AEs. There were no meaningful differences between treatment groups in vital signs, ECGs or clinical lab assessments.
In November 2016, we announced positive results from the second of our two pivotal clinical trials, evaluating pain relief over a 24-hour period in a soft tissue, post-operative pain model (abdominoplasty). In the trial, IV meloxicam achieved the primary endpoint of a statistically significant difference in SPID24, compared to placebo. In this multicenter, randomized, double-blind, placebo-controlled clinical trial, 219 patients were enrolled and randomly assigned to receive a postoperative regimen of IV meloxicam (30mg bolus injection) or placebo in a 1:1 ratio, once every 24 hours. The IV meloxicam treatment arm demonstrated a statistically significant reduction in SPID24 (p=0.0145) compared to the placebo arm. The study also achieved statistical significance for 10 of the secondary endpoints, including statistically significant differences in SPID12 (p=0.0434), time to perceptible pain relief (p=0.0050), subjects with ³30% improvement at 24 hours (p=0.0178), number of times patients required rescue in the first 24 hours after randomization (p=0.0275), as well as number of times rescued from 24 to 48 hours (p=0.0009), and several other pain relief metrics, compared to placebo. The safety results demonstrated that IV meloxicam was well tolerated with no difference in SAEs related to bleeding for IV meloxicam treated patients versus placebo (1 each). There were two additional SAEs observed in the placebo group. The most common (³2% in the IV meloxicam group) AEs were nausea, headache, vomiting, and dizziness. The incidence of these events was lower than those observed in the placebo group. The majority of AEs were mild in nature and one patient in the placebo group discontinued treatment due to an adverse event of post-procedural bleeding. There were no meaningful differences between treatment groups in vital signs, ECGs or clinical lab assessments.
2
To complete our Phase III program, we are currently enrolling patients following a variety of surgical conditions in an additional safety study of IV meloxicam. The population selected for inclusion in the safety study is intended to replicate real world use of injectable meloxicam. Overall we expect to enroll a total of approximately 1,100 patients in our Phase III program. If we continue to observe a favorable safety profile in our additional safety studies, we anticipate filing an NDA for IV meloxicam in the summer of 2017. We plan to pursue a Section 505(b)(2) regulatory strategy for IV meloxicam.
Recro Gainesville
Through our subsidiary, Recro Gainesville, we leverage our formulation and development expertise to develop and manufacture pharmaceutical products using our proprietary delivery technologies for commercial partners who commercialize or plan to commercialize these products. Our manufacturing and development capabilities include formulation through process development, scale-up and full-scale commercial manufacturing and specialized capabilities for the development and manufacturing of controlled substances. In a typical collaboration, we license certain intellectual property to our commercial partners and work with our commercial partners to develop product candidates, or new formulations of existing product candidates. In these collaborations, we also typically exclusively manufacture and supply clinical and commercial supplies of these product candidates. These collaborations result in revenue streams including from royalties, profit sharing, research and development and manufacturing, which support continued operations for Recro Gainesville as well as our research and development of proprietary product candidates. We currently develop and/or manufacture the following products with our commercial partners: Ritalin LA®, Focalin XR®, Verelan PM®, generic Verapamil and Zohydro ER®, as well as development stage products.
Our Pipeline Product Candidates
We also have a pipeline with other early-stage product candidates. Dex-IN, a proprietary intranasal formulation of dexmedetomidine, or Dex, is in a class of drugs called alpha-2 adrenergic agonists and is an FDA approved and commercial injectable drug, sold by Hospira, Inc. in the United States under the brand name Precedex® and by Orion Corporation, or Orion, in Europe under the brand name Dexdor®. We previously studied Dex-IN for the treatment of post-operative pain, but based on clinical trial results and feedback from the FDA, we are exploring other potential indications for Dex-IN, including for the treatment of peri-procedural pain. We also have a sublingual formulation of Dex, Dex-SL, which may be appropriate for use in treating chronic pain. In addition to Dex-IN and Dex-SL, we have another selective alpha-2 agonist product candidate in our pipeline, Fadolmidine, or Fado, which has been shown to be effective in a post-bunionectomy Phase II pain study conducted by Orion. Based on preclinical data, we believe Fado also shows promise in neuropathic pain.
Intellectual Property
We own patents and patent applications for injectable meloxicam, that cover compositions, including compositions produced using NanoCrystal® technology, method of making and method of treating. These issued patents expire in 2022 in the United States. We also in-license from Alkermes, on a perpetual, royalty-free basis, composition and methods of making patent and patent applications (specifically directed to the prevention of flake like substances) which expire in 2030.
We own various controlled release formulation patents, including patents in the United States, Canada, and Europe, related to our proprietary delivery technologies that we utilize in our drug development, formulation and manufacturing business through Recro Gainesville. These patents are scheduled to expire between 2019 and 2026. We own patents and patent applications in the United States and Canada directed to the composition of, manufacturing of, and formulating of Zohydro ER®. The patent protection for Zohydro ER® could provide for protection of Zohydro ER® through 2034, subject to extension.
3
We also hold patent applications directed to the analgesia indication, formulations and intranasal and transmucosal methods of use of Dex, and we are progressing through the patent application process globally, including the United States. Several patent applications have issued as patents outside the United States for transmucosal methods, and the resulting patent protection in the United States will last into 2030, subject to extension.
Our Strategy
We intend to maximize the value of our product candidates. Our strategy to achieve this goal includes:
| • | Advance IV meloxicam through clinical development and regulatory approval for moderate to severe pain. |
| • | Commercialize IV meloxicam in the United States independently or with third parties. |
| • | Expand our development, formulation and manufacturing business. |
| • | Enter into strategic partnerships to maximize the potential of our product candidates outside of the United States. |
| • | Leverage our management and development experience to explore other indications for injectable meloxicam and to develop our other pipeline product candidates. |
| • | Acquire additional products and product candidates. |
Financial Information
We have a limited operating history. In addition to revenue generated from Recro Gainesville, we have funded our operations to date primarily from proceeds received from public offerings and private placements of convertible preferred stock, convertible notes and common stock and our initial public offering of common stock, or IPO. On March 12, 2014, we closed our IPO in which we sold 4,312,500 shares of common stock for net proceeds of approximately $30.3 million. On July 7, 2015, we closed a Private Placement with certain accredited investors in which we sold 1,379,311 shares of common stock at a price per share of $11.60, for net proceeds of approximately $14.8 million. On August 19, 2016, we closed an underwritten public offering in which we sold 1,986,666 shares of common stock at a price per share of $7.50 for net proceeds of approximately $13.4 million. As of November 30, 2016, we have sold 1,143,940 shares of common stock under a common stock purchase agreement with Aspire Capital, LLC, or the Aspire Agreement, for net proceeds of approximately $7.8 million.
Corporate Information
We were incorporated under the laws of the Commonwealth of Pennsylvania in November 2007. Our principal executive offices are located at 490 Lapp Road, Malvern, PA 19355, and our telephone number is (484) 395-2470.
Available Information
Our website address is www.recropharma.com. The information contained in, or accessible through, our website does not constitute part of this prospectus. We make available free of charge on our website our annual, quarterly and current reports, including amendments to such reports, as soon as reasonably practicable after we electronically file such material with, or furnish such material to, the Securities and Exchange Commission, or the SEC. Information contained on our website is not incorporated by reference into this prospectus, and you should not consider information contained on our website as part of this prospectus.
4
Risks Associated With Our Business
Our business is subject to a number of risks of which you should be aware before making an investment decision. These risks are discussed more fully in the “Risk Factors” section of this prospectus immediately following this prospectus summary and in Part I, Item 1A “Risk Factors” of our Annual Report on Form 10-K filed with the SEC on March 24, 2016, which is incorporated by reference in this prospectus. These risks include the following:
| • | We have incurred significant losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future. |
| • | We depend substantially on the successful completion of our Phase III clinical trial program for injectable meloxicam. The positive clinical results obtained for injectable meloxicam in earlier clinical studies may not be repeated in our remaining Phase III safety study and, thus, we may never receive regulatory approval of injectable meloxicam. |
| • | We have only recently begun to generate revenue through our acquisition of our contract manufacturing facility, royalty and formulation business, but we may never be profitable. |
| • | Revenues from our manufacturing business are dependent on a small number of commercial partners, and the loss of one of these partners, or a decline in their orders, may adversely affect our business. Our four largest customers generated 96% of our revenues for the nine months ended September 30, 2016, of which one customer generated 43% of our revenue, and another customer generated 38% of our revenue under two separate customer agreements. |
| • | We depend substantially on the successful completion of clinical trials for our other product candidates. The positive results obtained for these other product candidates in earlier pre-clinical and clinical studies may not be repeated and, thus, we may never receive regulatory approval of these other product candidates. |
| • | Even if we obtain FDA approval for injectable meloxicam or our other product candidates in the United States, we may never obtain approval for or commercialize our products outside of the United States, which would limit our ability to realize their full market potential. |
| • | We have incurred substantial indebtedness, which could adversely affect our business. |
| • | We use third parties to assist with conducting, supervising and monitoring portions of our clinical studies, and if those third parties perform in an unsatisfactory manner, it may harm our business. |
| • | We are subject to intense competition and, if we are unable to compete effectively, our product candidates may not reach their commercial potential. |
Implications of Being an Emerging Growth Company
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. We will remain an emerging growth company until the earliest of (1) the beginning of the first fiscal year following the fifth anniversary of our initial public offering, or January 1, 2020, (2) the beginning of the first fiscal year after our annual gross revenue is $1.0 billion or more, (3) the date on which we have, during the previous three-year period, issued more than $1.0 billion in non-convertible debt securities and (4) as of the end of any fiscal year in which the market value of our common stock held by non-affiliates exceeded $700 million as of the end of the second quarter of that fiscal year.
For as long as we remain an “emerging growth company,” we may take advantage of certain exemptions from various reporting requirements that are applicable to public companies that are not “emerging
5
growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation and financial statements in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote to approve executive compensation and shareholder approval of any golden parachute payments not previously approved. We will take advantage of these reporting exemptions until we are no longer an “emerging growth company.”
The JOBS Act provides that an “emerging growth company” can take advantage of an extended transition period for complying with new or revised accounting standards. We have irrevocably elected not to avail ourselves of this exemption and, therefore, we are subject to the same new or revised accounting standards as other public companies that are not “emerging growth companies.”
Summary Financial Data
We derived the consolidated statements of operations data presented below for the years ended December 31, 2015 and 2014 from our audited financial statements. The consolidated statements of operations data for the nine months ended September 30, 2016 and 2015, and the consolidated balance sheet data as of September 30, 2016, are derived from our unaudited interim financial statements. We have prepared the unaudited interim financial statements on the same basis as the audited financial statements and have included, in our opinion, all adjustments, consisting only of normal recurring adjustments that we consider necessary for a fair presentation of the financial information set forth in those statements. Our historical results are not necessarily indicative of the results that should be expected in the future, and our interim results are not necessarily indicative of the results that should be expected for the full year or any other period. The following information should be read in conjunction with our consolidated financial statements and related notes incorporated by reference in this prospectus from our Annual Report on Form 10-K for the fiscal year ended December 31, 2015 and our Quarterly Report on Form 10-Q for the quarter ended September 30, 2016.
The as adjusted balance sheet data as of September 30, 2016 reflects receipt of the estimated net proceeds of $ million from the sale of the common stock in this offering at the public offering price of $ per share, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
6
| Year ended December 31, | Nine months ended September 30, |
|||||||||||||||
| 2015 | 2014 | 2016 | 2015 | |||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||
| Statements of Operations Data: |
||||||||||||||||
| Revenue: |
||||||||||||||||
| Manufacturing, royalty and profit sharing revenue |
$ | 49,284 | $ | — | $ | 50,260 | $ | 32,824 | ||||||||
| Research and development revenue |
2,668 | — | 1,713 | 2,375 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total revenue |
51,952 | — | 51,973 | 35,199 | ||||||||||||
| Operating expenses: |
||||||||||||||||
| Cost of sales (excluding amortization of intangible assets) |
28,054 | — | 25,563 | 19,228 | ||||||||||||
| Research and development |
12,281 | 7,874 | 23,175 | 7,260 | ||||||||||||
| General and administrative |
13,017 | 3,998 | 9,263 | 8,492 | ||||||||||||
| Amortization of intangible assets |
1,884 | — | 1,937 | 1,238 | ||||||||||||
| Change in warrant valuation |
(1,560 | ) | — | 47 | 119 | |||||||||||
| Change in contingent consideration valuation |
5,246 | — | 7,705 | 2,586 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expenses |
58,922 | 11,872 | 67,690 | 38,923 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating loss |
(6,970 | ) | (11,872 | ) | (15,717 | ) | (3,724 | ) | ||||||||
| Other income (expense): |
||||||||||||||||
| Interest income |
12 | 11 | 27 | 10 | ||||||||||||
| Interest expense |
(5,560 | ) | (4,273 | ) | (4,279 | ) | (3,888 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss before income taxes |
(12,518 | ) | (16,134 | ) | (19,969 | ) | (7,602 | ) | ||||||||
| Income tax benefit |
15,551 | — | 166 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net income (loss) |
3,033 | (16,134 | ) | (19,803 | ) | (7,602 | ) | |||||||||
| Accretion of redeemable convertible preferred stock |
— | (1,270 | ) | — | — | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net income (loss) applicable to common shareholders |
3,033 | (17,404 | ) | (19,803 | ) | (7,602 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Basic net income (loss) per common share |
$ | 0.36 | $ | (2.79 | ) | $ | (2.01 | ) | $ | (0.92 | ) | |||||
|
|
|
|
|
|
|
|
|
|||||||||
| Diluted net income (loss) per common share |
$ | 0.21 | $ | (2.79 | ) | (2.01 | ) | (0.92 | ) | |||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Weighted average basic common share outstanding |
8,491,025 | 6,238,581 | 9,862,526 | 8,243,909 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Weighted average diluted common share outstanding |
8,749,234 | 6,238,581 | 9,862,526 | 8,243,909 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| As of September 30, 2016 | ||||||||||||
| Actual | Pro Forma(1) | As Adjusted(2) | ||||||||||
| (in thousands) | ||||||||||||
| Balance Sheet Data: |
||||||||||||
| Cash and cash equivalents |
$ | 24,752 | $ | 28,373 | ||||||||
| Working capital |
37,736 | 41,357 | ||||||||||
| Total assets |
146,009 | 149,630 | ||||||||||
| Debt (including current portion)(3) |
24,236 | 24,236 | ||||||||||
| Total shareholders’ equity |
40,631 | 44,252 | ||||||||||
| (1) | The pro forma column reflects the sale of 500,000 shares of common stock for approximately $3.6 million in net proceeds under the Aspire Agreement between October 1, 2016 and November 22, 2016. |
| (2) | As adjusted to reflect the sale of the shares being offered in this offering, and the receipt of the estimated net proceeds of $ million from the sale of these shares, at the public offering price of $ per share and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. |
| (3) | Includes principal balance outstanding of $27,347, net of unamortized deferred issuance costs of $3,111. |
7
The Offering
| Common stock offered by us |
shares ( shares if the underwriters’ option to purchase additional shares is exercised in full). | |
| Common stock to be outstanding after this offering |
shares ( shares if the underwriters’ option to purchase additional shares is exercised in full). | |
| Option to purchase additional shares |
The underwriters have the option to purchase from us up to a maximum of additional shares of common stock. The underwriters can exercise this option at any time within 30 days from the date of this prospectus. | |
| Use of proceeds |
We estimate that the net proceeds to us from this offering, after deducting underwriting discounts and commissions and estimated offering expenses payable by us, will be approximately $ million. We intend to use the net proceeds from this offering to fund the NDA filing and regulatory approval process and the initiation of commercial activities for IV meloxicam, our planned IV meloxicam Phase IIIB program, investments in our pipeline product candidates, and for general corporate purposes. See “Use of Proceeds.” | |
| Risk Factors |
An investment in our common stock involves a high degree of risk. See “Risk Factors” beginning on page 8 of this prospectus and the similarly titled sections in the documents incorporated by reference into this prospectus . | |
| NASDAQ Capital Market symbol |
REPH | |
8
Outstanding Shares
The number of shares of our common stock to be outstanding after this offering is based on 11,863,660 shares of our common stock outstanding as of September 30, 2016, and excludes:
| • | 2,343,819 shares of our common stock issuable upon the exercise of stock options outstanding as of September 30, 2016 at a weighted-average exercise price of $7.00 per share; |
| • | 19,975 shares of our common stock issuable upon the vesting and settlement of restricted stock units outstanding as of September 30, 2016; |
| • | 174 shares of our common stock available for future issuance as of September 30, 2016 under our 2008 Stock Option Plan; |
| • | 855,022 shares of our common stock available for future issuance as of September 30, 2016 under our Amended and Restated Equity Incentive Plan; |
| • | 784,928 shares of our common stock issuable upon the exercise of outstanding warrants as of September 30, 2016 with a weighted average exercise price of $12.05 per share; and |
| • | 500,000 shares of our common stock sold from October 1, 2016 to November 22, 2016 under the Aspire Agreement. |
Except as otherwise indicated herein, all information in this prospectus, including the number of shares that will be outstanding after this offering, does not assume or give effect to the exercise of options or warrants outstanding as of September 30, 2016.
9
An investment in our common stock involves a high degree of risk. Before deciding whether to invest in our common stock, you should carefully consider the risks described below and those discussed under the Section captioned “Risk Factors” contained in our Annual Report on Form 10-K for the fiscal year ended December 31, 2015, which is incorporated by reference in this prospectus, together with the information included in this prospectus and documents incorporated by reference herein, and in any free writing prospectus that we have authorized for use in connection with this offering. If any of these risks actually occurs, our business, financial condition, results of operations or cash flow could be harmed. This could cause the trading price of our common stock to decline, resulting in a loss of all or part of your investment.
Risks Related to This Offering
Management will have broad discretion as to the use of the proceeds from this offering, and we may not use the proceeds effectively.
Our management will have broad discretion with respect to the use of proceeds of this offering, including for any of the purposes described in the section of this prospectus entitled “Use of Proceeds.” You will be relying on the judgment of our management regarding the application of the proceeds of this offering. The results and effectiveness of the use of proceeds are uncertain, and we could spend the proceeds in ways that you do not agree with or that do not improve our results of operations or enhance the value of our common stock. Our failure to apply these funds effectively could harm our business, delay the development of our product candidates and cause the price of our common stock to decline.
You will experience immediate and substantial dilution in the net tangible book value per share of the common stock you purchase.
Since the public offering price for our common stock in this offering is substantially higher than the net tangible book value per share of our common stock outstanding prior to this offering, you will suffer immediate and substantial dilution in the net tangible book value of the common stock you purchase in this offering. See the section titled “Dilution” below for a more detailed discussion of the dilution you will incur if you purchase shares in this offering.
Issuances of shares of common stock or securities convertible into or exercisable for shares of common stock following this offering, as well as the exercise of options and warrants outstanding, will dilute your ownership interests and may adversely affect the future market price of our common stock.
The issuance of additional shares of our common stock could be dilutive to shareholders if they do not invest in future offerings. We intend to use the net proceeds from this offering to fund the NDA filing and regulatory approval process and the initiation of commercial activities for IV meloxicam, our planned IV meloxicam Phase IIIB program, investments in our pipeline product candidates, and for general corporate purposes. We will need additional capital to fund the completion of our commercial infrastructure, including milestone payments, and post-launch activities for IV meloxicam, subject to final regulatory approval. We may seek additional capital through a combination of private and public equity offerings, debt financings, strategic partnerships and alliances and licensing arrangements, which may cause your ownership interest to be diluted.
In addition, we have a significant number of options and warrants to purchase shares of our common stock outstanding. If these securities are exercised, you may incur further dilution. Moreover, to the extent that we issue additional options or warrants to purchase, or securities convertible into or exchangeable for, shares of our common stock in the future and those options, warrants or other securities are exercised, converted or exchanged, shareholders may experience further dilution.
10
A significant portion of our total outstanding shares are eligible to be sold into the market, which could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market, either by us or by our current shareholders, or the perception that these sales could occur, could cause a decline in the market price of our securities. Such sales, along with any other market transactions, could adversely affect the market price of our common stock.
Upon completion of this offering, based on our shares outstanding as of December , 2016, we will have shares of common stock outstanding. Of these shares, only are subject to a contractual lock-up with the underwriters for this offering for a period of 90 days following this offering. These shares can be sold, subject to any applicable volume limitations under federal securities laws, after the earlier of the expiration of, or release from, the 90-day lock-up period. The balance of our outstanding shares of common stock, including any shares purchased in this offering, may be resold into the public market immediately without restriction, unless owned or purchased by our affiliates. Moreover, some of the holders of our common stock have the right, subject to specified conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders.
As of November 30, 2016, there were approximately 2,407,681 shares subject to outstanding options and restricted stock unit awards or that are otherwise issuable under our equity compensation plans, all of which shares we have registered under the Securities Act of 1933, as amended, on a registration statement on Form S-8. These shares can be freely sold in the public market upon issuance, subject to volume limitations applicable to affiliates and the lock-up agreements described above, to the extent applicable.
11
This prospectus and the documents incorporated by reference herein contain forward-looking statements that involve substantial risks and uncertainties. All statements, other than statements of historical facts, included in this prospectus or the documents incorporated herein by reference regarding our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans and objectives of management are forward-looking statements. The words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “plan,” “predict,” “project,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward- looking statements contain these identifying words.
The forward-looking statements in this prospectus and the documents incorporated herein by reference include, among other things, statements about:
| • | the results and timing of our clinical trials of injectable meloxicam or our other product candidates, and any future clinical and preclinical studies; |
| • | unfavorable new clinical data and additional analyses of existing clinical data; |
| • | whether results of early clinical trials will be indicative of the results of future clinical trials and whether interim results from a clinical trial will be predictive of the final results of the clinical trial; |
| • | the ability to obtain and maintain regulatory approval of our product candidates, and the labeling under any approval that we may obtain; |
| • | regulatory developments in the United States and foreign countries; |
| • | our plans to develop and commercialize our product candidates; |
| • | our ability to raise future financing for continued development; |
| • | the performance of our third-party suppliers and manufacturers; |
| • | our ability to obtain patent protection and defend our intellectual property rights; |
| • | our ability to successfully implement our strategy; |
| • | our ability to maintain our relationships and contracts with our commercial partners; |
| • | our ability to comply with stringent U.S. and foreign government regulation in the manufacture of pharmaceutical products, including Good Manufacturing Practice, or cGMP, compliance and U.S. Drug Enforcement Agency, or DEA, compliance; and |
| • | our ability to meet required debt payments, including any milestone payments owing to Alkermes plc, and operate under increased leverage and associated lending covenants. |
We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. We have included important factors in the cautionary statements included in this prospectus, particularly under “Risk Factors,” that we believe could cause actual results or events to differ materially from the forward-looking statements that we make. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures, collaborations or investments we may make.
You should read this prospectus and the documents that we incorporate by reference herein completely and with the understanding that our actual future results may be materially different from what we expect. We do not assume any obligation to update any forward-looking statements.
12
We estimate that the net proceeds from our issuance and sale of shares of common stock in this offering will be $ million, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
We intend to use the net proceeds from this offering to fund the NDA filing and regulatory approval process and the initiation of commercial activities for IV meloxicam, our planned IV meloxicam Phase IIIB program, investments in our pipeline product candidates, and for general corporate purposes. We will need additional capital to fund the completion of our commercial infrastructure, including milestone payments, and post-launch activities for IV meloxicam, subject to regulatory approval.
The expected use of the net proceeds from this offering represents our intentions based upon our current plans and business conditions, which could change in the future as our plans and business conditions evolve. The amounts and timing of our actual expenditures may vary significantly depending on numerous factors, including our ability to gain access to additional financing, the relative success and cost of our research, preclinical and clinical development programs and whether we are able to enter into future licensing or collaboration arrangements. As a result, our management will have broad discretion in the application of the net proceeds from this offering, and investors will be relying on the judgment of our management regarding the application of the net proceeds from this offering. Pending their ultimate use, we intend to invest the net proceeds from this offering in short-term, investment-grade, interest-bearing securities.
13
MARKET PRICE OF OUR COMMON STOCK
Our common stock has been listed on the Nasdaq Capital Market under the symbol “REPH” since our initial public offering on March 12, 2014. Prior to that time, there was no public market for our common stock. The following table sets forth the high and low sale prices per share for our common stock on the Nasdaq Capital Market for the periods indicated:
| HIGH | LOW | |||||||
| 2016 |
||||||||
| First Quarter |
$ | 9.20 | $ | 5.59 | ||||
| Second Quarter |
$ | 8.62 | $ | 5.95 | ||||
| Third Quarter |
$ | 12.50 | $ | 7.51 | ||||
| Fourth Quarter (through November 30, 2016) |
$ | 10.17 | $ | 6.89 | ||||
| 2015 |
||||||||
| First Quarter |
$ | 9.93 | $ | 2.80 | ||||
| Second Quarter |
$ | 15.40 | $ | 6.56 | ||||
| Third Quarter |
$ | 18.30 | $ | 11.06 | ||||
| Fourth Quarter |
$ | 12.86 | $ | 7.58 | ||||
| 2014 |
||||||||
| First Quarter (beginning March 12, 2014) |
$ | 9.88 | $ | 7.00 | ||||
| Second Quarter |
$ | 8.49 | $ | 5.01 | ||||
| Third Quarter |
$ | 8.10 | $ | 2.71 | ||||
| Fourth Quarter |
$ | 3.39 | $ | 2.36 | ||||
On November 30, 2016, the closing price of our common stock as reported by the Nasdaq Capital Market was $8.00 per share. As of November 30, 2016, there were approximately 9 holders of record of our common stock. We believe that the number of beneficial owners of our common stock at that date was substantially greater.
14
We have never declared or paid any cash dividends on our common stock and our ability to pay cash dividends is currently prohibited by the terms of our credit facility with OrbiMed. We do not anticipate paying cash dividends on our common stock in the foreseeable future. Payment of future dividends, if any, on our common stock will be at the discretion of our board of directors after taking into account various factors, including our financial condition, operating results, anticipated cash needs and plans for expansion.
15
Overview
We are a revenue-generating, specialty pharmaceutical company primarily focused on developing innovative products for hospitals and ambulatory care settings. Our lead product candidate is a proprietary injectable form of meloxicam, a long-acting preferential COX-2 inhibitor, the oral form of which has been marketed by Boehringer Ingelheim Pharmaceuticals, Inc. since the 1990s as Mobic®. IV meloxicam has successfully completed five clinical trials in the treatment of moderate to severe pain, including a two recently completed pivotal Phase III clinical trials, one evaluating pain relief over a 48-hour period in a hard tissue, post-operative pain model (bunionectomy) and the other evaluating pain relief over a 24-hour period in a soft tissue, post-operative pain model (abdominoplasty). We believe that IV meloxicam compares favorably to competitive therapies in onset of pain relief, duration of pain relief, extent of pain relief and time to peak analgesic effect. To complete our Phase III program, we are currently enrolling patients following a variety of surgical conditions in an additional safety study of IV meloxicam. The population selected for inclusion in the safety study is intended to replicate real world use of injectable meloxicam. Overall we expect to enroll a total of approximately 1,100 patients in our Phase III program. If we continue to observe a favorable safety profile in our additional safety studies, we anticipate filing an NDA for IV meloxicam to the FDA in the summer of 2017. As a non-opioid product, we believe injectable meloxicam will overcome many of the issues associated with commonly prescribed opioid therapeutics, including addiction, misuse/diversion, respiratory depression and constipation while maintaining analgesic, or pain relieving, effects. We are pursuing a Section 505(b)(2) regulatory strategy for injectable meloxicam.
In addition to developing proprietary drug candidates, we, through our subsidiary Recro Gainesville, leverage our formulation expertise to develop and manufacture pharmaceutical products using our proprietary delivery technologies for commercial partners who commercialize or plan to commercialize these products. These collaborations result in revenue streams including royalties, profit sharing, research and development and manufacturing, which support continued operations for Recro Gainesville as well as our research and development of proprietary product candidates. Recro Gainesville operates a 97,000 square foot, DEA-licensed manufacturing facility in Gainesville, Georgia. We currently develop and/or manufacture the following products with our commercial partners: Ritalin LA®, Focalin XR®, Verelan PM®, generic Verapamil and Zohydro ER®, as well as development stage products.
Our pipeline also includes other early-stage product candidates. Dex-IN, a proprietary intranasal formulation of dexmedetomidine, or Dex, is in a class of drugs called alpha-2 adrenergic agonists, and is an FDA approved and commercial injectable drug, sold by Hospira, Inc. in the United States under the brand name Precedex® and by Orion in Europe under the brand name Dexdor®. We previously studied Dex-IN for the treatment of post-operative pain, but based on clinical trial results and feedback from the FDA, we are exploring other potential indications for Dex-IN, including for the treatment of peri-procedural pain. We also have a sublingual formulation of Dex, Dex-SL, which may be appropriate for use in treating chronic pain. In addition to Dex-IN and Dex-SL, we have another selective alpha-2 agonist product candidate in our pipeline, Fadolmidine, or Fado, which we believe also shows promise in neuropathic pain based on preclinical data.
16
Pipeline

Corporate History and Significant Milestones
| • | We were incorporated in 2007, and began operating in 2008. |
| • | In March 2014, we closed our IPO in which we sold 4,312,500 shares of common stock for net proceeds of approximately $30.3 million. |
| • | In February 2015, we entered into a common stock purchase agreement with Aspire Capital Fund, LLC, or Aspire, under which, as of November 30, 2016, we have sold 1,143,940 shares of common stock for net proceeds of approximately $7.8 million. |
| • | In April 2015, we completed a transformative acquisition in which we acquired from Alkermes plc, or Alkermes, certain assets, including the worldwide rights to injectable meloxicam and our development, formulation and manufacturing business now operating through Recro Gainesville, which we refer to herein as the Gainesville Transaction. This transaction transformed our business through the addition of a revenue-generating business and the increase in our workforce as a result of the addition of the Recro Gainesville employees. The consideration paid consisted of $50.0 million, a $4.0 million working capital adjustment and a seven-year warrant to purchase 350,000 shares of our common stock at an exercise price of $19.46 per share. In addition, we may be required to pay up to an additional $120.0 million in milestone payments (including $10 million payable upon NDA filing, $30 million payable upon regulatory approval and additional payments upon net sales milestones) and a percentage of future product net sales related to injectable meloxicam. |
| • | In April 2015, to fund the up-front payment due to Alkermes, we entered into a credit agreement with OrbiMed Royalty Opportunities II, LP, or OrbiMed. The interest rate under the credit agreement is equal to LIBOR plus 14.0%, with a 1.0% LIBOR floor. Pursuant to the credit agreement, we issued OrbiMed a warrant to purchase an aggregate of 294,928 shares of our common stock at an exercise price of $3.28 per share, subject to certain adjustments. As of September 30, 2016, we have paid down approximately $22.7 million, or 45%, of the original $50.0 million of the senior secured term loan from the business’s excess cash flow generated. |
17
| • | In July 2015, we completed a private placement with certain accredited investors in which we sold 1,379,311 shares of common stock at a price per share of $11.60, for net proceeds of approximately $14.8 million. |
| • | In July 2016, we announced positive results from a Phase III pivotal clinical trial evaluating our lead product candidate, injectable meloxicam, in pain relief over a 48-hour period in a hard tissue, post-operative pain model. |
| • | In August 2016, we closed an underwritten public offering in which we sold 1,986,666 shares of common stock at a price per share of $7.50, for net proceeds of approximately $13.4 million after deducting underwriting commissions and offering expenses. |
| • | In November 2016, we announced positive results from the second of our Phase III pivotal clinical trial evaluating our lead product candidate, injectable meloxicam, in pain relief over a 24-hour period in a soft tissue, post-operative pain model. |
Our Strategy
We intend to maximize the value of our product candidates. This strategy could include developing our candidates through approval and ultimately self-commercialization, out-licensing, partnering on certain assets, or selling the Company or the assets. Our broader corporate strategy includes the following:
Advance IV meloxicam through clinical development and regulatory approval for moderate to severe pain. Our key goal is to file an NDA, and receive FDA approval of IV meloxicam for the management of moderate to severe pain as soon as possible. IV meloxicam has recently successfully completed two pivotal Phase III clinical trials. To complete our Phase III program, we are currently enrolling patients following a variety of surgical conditions in an additional safety study of IV meloxicam. If we continue to observe a favorable safety profile in our additional safety studies, we anticipate filing the NDA for IV meloxicam with the FDA in the summer of 2017.
Commercialize IV meloxicam in the United States independently or with third parties. We believe IV meloxicam targets a narrow group of specialist prescribers which would allow for the successful marketing and commercialization by a company of our size. We are currently preparing for a potential U.S. commercial launch of IV meloxicam, if approved, and we plan to establish sales, marketing and reimbursement functions to commercialize IV meloxicam in the United States.
Expand our development, formulation and manufacturing business. We are focused on expanding our development, formulation and manufacturing services. We intend to seek additional manufacturing and development partnerships with commercial partners through ongoing business development efforts, as well as through expansion of our proprietary drug delivery technologies, and service offerings.
Enter into strategic partnerships to maximize the potential of our product candidates outside of the United States. We intend to pursue strategic collaborations with other pharmaceutical companies to develop and commercialize our product candidates outside of the United States. We believe that our management expertise and unique product candidates make us an attractive partner to potential strategic companies.
Leverage our management and development experience to explore other indications for injectable meloxicam and to develop our other pipeline product candidates. If we have sufficient additional resources, we plan to evaluate injectable meloxicam for potential additional indications. In addition, our early-stage product pipeline includes proprietary drug solutions for peri-procedural pain and pain resulting from cancer, musculoskeletal disorders and peripheral neuropathy, utilizing multiple delivery systems, including intrathecal/epidural, transdermal, intranasal and sublingual platforms. Our goal is to
18
leverage our drug development expertise along with innovative delivery systems to develop these product candidates to improve quality of life for the millions of people suffering from moderate-to-severe pain annually.
Acquire additional products and product candidates. We may identify and license, co-promote or acquire products or product candidates for development for use in hospital or ambulatory care settings.
Our Lead Product Candidate – Injectable Meloxicam
Meloxicam is a long-acting, preferential COX-2 inhibitor that possesses anti-inflammatory, analgesic, and antipyretic activities, which are believed to be related to the inhibition of cyclooxygenase, or COX, and subsequent reduction in prostaglandin biosynthesis. Meloxicam has been marketed by Boehringer Ingelheim Pharmaceuticals, Inc. since the 1990s as an oral agent, Mobic®. Mobic tablets and suspension are indicated for the relief of the signs and symptoms of osteoarthritis and rheumatoid arthritis and the relief of the signs and symptoms of pauciarticular or polyarticular juvenile rheumatoid arthritis in patients 2 years or older.
Meloxicam has a slow onset of action orally, and is not currently approved for the treatment of acute pain. The oral form has a prolonged absorption time, with the time of maximum observed plasma concentration, or tmax, being approximately 5-6 hours following oral administration, which is consistent with its poor aqueous solubility. Our proprietary injectable form of the drug, which utilizes NanoCrystal™ technology, provides a faster onset of action of meloxicam, thus providing a rapid and sustained treatment of acute pain via the IV or intramuscular, or IM, administration routes.
Post-Operative Pain Market
Based upon statistics from the National Center for Health Statistics, it is estimated that there are over 100 million surgeries performed in the United States each year. Of these surgeries, we believe at least 50 million procedures require post-operative pain medication. While opioids are generally considered the most effective treatment for post-operative pain, they raise serious concerns due to addiction, illicit use, respiratory depression and other side effects, including constipation, nausea, vomiting and intolerance. Due to their addictive potential, opioids are regulated as controlled substances and are listed on Schedule II and III by the DEA. As a result of these side effects, pain sufferers tend to limit their use of opioids, resulting in as many as 40% of post-operative patients reporting inadequate pain relief. This reduces the quality of life for individuals and creates an economic burden estimated to be at least $560 to $635 billion a year in medical costs and lost productivity. According to a January 2016 article in the New England Journal of Medicine, overdose deaths from prescription painkillers (defined to mean opioid or narcotic pain relievers) have increased significantly over the past 14 years. It notes the following trends:
| • | Prescription painkiller overdoses killed 18,893 people in the United States in 2014; |
| • | In 2014, about 10.3 million Americans (age 12 or older) reported nonmedical use of prescription painkillers in the past year; and |
| • | Emergency department visits involved with misusing or abusing prescription opioid painkillers increased 153% between 2004 and 2011. |
We believe that injectable meloxicam offers an attractive alternative for relief of moderate to severe pain without the risks associated with opioids, and we believe that the majority of patients to be served would be in the post-operative setting. Accordingly, we believe that physicians and third-party payors, including Medicare and Medicaid, are highly interested in new non-opioid pain therapies that provide effective post-operative pain relief without the adverse issues associated with opioids.
19
Injectable Meloxicam Advantages
We believe injectable meloxicam has a number of advantages over existing, FDA approved analgesics, including the following:
Not considered a controlled substance. Meloxicam is not an opioid and not a controlled substance. Opioid therapeutics are currently controlled by the DEA under the Controlled Substances Act. Under this act, opioids have been scheduled based on their potential for abuse and/or addiction. For those opioids placed in Schedule II, federal law prohibits the refilling of prescriptions, thus requiring patients to request, and physicians to write, additional prescriptions for each refill. Examples of Schedule II opioids include codeine, fentanyl, sufentanil, hydrocodone and oxycodone.
Does not cause respiratory depression. Meloxicam does not cause respiratory depression. Besides the addictive nature of opioids, we believe that medical practitioners are highly concerned with respiratory depression, which is a well-documented side effect of opioid use (all opioids, including fentanyl and oxycodone). Respiratory depression, which is defined by inadequate ventilation leading to increased carbon dioxide levels and respiratory acidosis, is an established outcome of opioid use. One of the more concerning adverse effects of chronic opioid use, for which tolerance does not develop, is respiratory depression during sleep, which can be life threatening. Meloxicam has demonstrated through multiple clinical trials and patient use that it does not cause respiratory depression.
Onset of pain relief. While the oral form of meloxicam can take 60 minutes or more for pain relief, the utilization of NanoCrystalTM technology in the IV formulation results in a more rapid onset of pain relief of less than 10 minutes. Ketorolac, for example, can take up to 30 minutes for the onset of pain relief.
Duration of pain relief. IV meloxicam utilizing NanoCrystalTM technology has demonstrated the potential to be an effective analgesic for up to 18 to 24 hours after a single dose in clinical trials. IV forms of ketorolac, ibuprofen and acetaminophen provide effective pain relief up to four to six hours, resulting in the need for four to six doses for every 24 hours.
Time to peak analgesic effect. Clinical data has demonstrated that IV meloxicam reaches peak analgesic effect within approximately 40 minutes of administration, reaching its peak faster than competing non-opioid therapeutics. Ketorolac can take between 1 to 2 hours to reach its peak analgesic effect.
Administration. We believe that IV meloxicam has an administration advantage in terms of bolus injection, whereas ibuprofen and acetaminophen can take up to 15 to 30 minutes to infuse. In addition, there is an IM formulation of meloxicam, while neither ibuprofen nor acetaminophen currently have IM formulations.
Clinical Development
Multiple clinical trials have been conducted to evaluate the safety, pharmacokinetics and analgesic potential of IV meloxicam. Based on the results of these trials, we believe IV meloxicam has the potential to be a potent analgesic in the management of moderate to severe pain. In early 2016, based on feedback from the FDA, we commenced our Phase III clinical trial program for IV meloxicam. The program includes two pivotal Phase III clinical trials, both of which IV meloxicam has successfully completed. To complete our Phase III program, we are currently enrolling patients following a variety of surgical conditions in an additional safety study of IV meloxicam. The population selected for inclusion in the safety study is intended to replicate real world use of injectable meloxicam. Overall we expect to enroll a total of approximately 1,100 patients in our Phase III program. If we continue to observe a favorable safety profile in our additional safety study, we anticipate filing an NDA for IV meloxicam in the summer of 2017.
20
Phase III Clinical Trials
Study REC-15-016
In July 2016 we announced positive results from one pivotal clinical trial, evaluating pain relief over a 48-hour period in a hard tissue, post-operative pain model (bunionectomy). In the trial, IV meloxicam achieved the primary endpoint of a statistically significant difference in SPID over the first 48 hours, or SPID48, compared to placebo. This was a Phase III, randomized, multicenter, multi-dose, double-blind, placebo-controlled study evaluating IV meloxicam in the management of post-operative pain following bunionectomy surgery. Two hundred and one patients who met the eligibility criteria were randomized to receive either IV meloxicam (30 mg) or placebo once daily for three days. Following the beginning of treatment, patients remained under observation for 48 hours at study centers. Patients were followed for 7 days after the initial dose of study medication. There was an oral opioid rescue treatment available to all patients, if required. The primary objective of the trial was to evaluate pain relief over a 48-hour period of IV meloxicam when administered as a bolus injection (over 15-30 seconds).
The primary efficacy endpoint of the trial was SPID48, utilizing a windowed 2-hour last observation carried forward, or W2LOCF, analysis method. Secondary efficacy endpoints included use of opioid rescue medication, SPIDs over various time intervals, and patient global assessment, or PGA, of pain control. The IV meloxicam treatment arm demonstrated a statistically significant reduction in SPID48 (p=0.0034) compared to the placebo arm (Figure 1).
Figure 1: SPID48
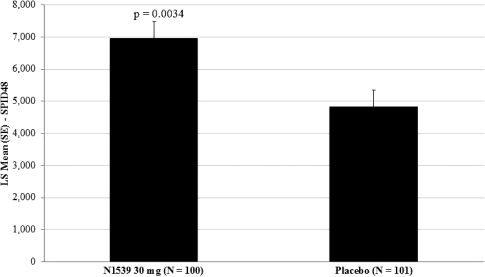
The study also achieved 15 secondary endpoints, including statistically significant differences in SPID6 (p=0.0153), SPID12 (p=0.0053), SPID24 (p=0.0084), SPID24-48 (p=0.0050), time to first use of rescue medication (p=0.0076), and several other rescue use and pain relief metrics during the first 48 hours, compared to placebo (Table 1).
21
Table 1: Summary of Secondary Endpoints
| Parameter |
p-value | |||
| SPID6 |
0.0153 | |||
| SPID12 |
0.0053 | |||
| SPID24 |
0.0084 | |||
| SPID24-48 |
0.0050 | |||
| Time to First Rescue Analgesia |
0.0076 | |||
| Number of Subjects Rescued 0-24 Hours |
0.0002 | |||
| Number of Subjects Rescued 24-48 Hours |
0.0009 | |||
| Number of Subjects Rescued 0-48 Hours |
0.0002 | |||
| Number of Times Rescued 0-24 Hours |
0.0025 | |||
| Number of Times Rescued 24-48 Hours |
0.0108 | |||
| Number of Times Rescued 0-48 Hours |
0.0014 | |||
| % Subjects with >30% Improvement – 6 Hours |
0.0451 | |||
| % Subjects with >30% Improvement – 24 Hours |
0.0107 | |||
| % Subjects with >50% Improvement – 24 Hours |
0.0430 | |||
| PGA of Pain Control at 48 hours |
0.0046 | |||
Times to Perceptible and Meaningful Pain Relief, % Subjects with >50% Improvement within 6 Hours, PGA of Pain Control at 24 hours were not significantly different between treatment groups.
The safety results demonstrated that IV meloxicam was well tolerated with no SAEs or bleeding events in the IV meloxicam-treated patients. The most common AEs occurring in at least 3% of IV meloxicam-treated treated patients, were nausea, headache, pruritus, constipation vomiting, dizziness, flushing and somnolence, and were comparable to the placebo group (Table 2). The IV meloxicam-treated patients experienced injection site pain and injection site erythema at a rate comparable to placebo. The majority of treatment emergent AEs, or TEAEs, were mild in nature and there were no discontinuations due to AEs. There were no meaningful differences between treatment groups in vital signs, ECGs or clinical lab assessments.
Table 2: Adverse Events reported by ³3% of subjects from any treatment group
| n (%) of Subjects | ||||
| Preferred Term |
N1539 30 mg (N=100) |
Placebo (N=101) | ||
| Subjects with ³1 TEAE |
44 (44.0) | 54 (53.5) | ||
| Nausea |
20 (20.0) | 26 (25.7) | ||
| Headache |
8 (8.0) | 12 (11.9) | ||
| Vomiting |
3 (3.0) | 9 (8.9) | ||
| Pruritus |
8 (8.0) | 3 (3.0) | ||
| Decreased appetite |
2 (2.0) | 7 (6.9) | ||
| Constipation |
4 (4.0) | 5 (5.0) | ||
| Abdominal pain |
— | 6 (5.9) | ||
| Dizziness |
3 (3.0) | 4 (4.0) | ||
| Flushing |
3 (3.0) | 1 (1.0) | ||
| Somnolence |
3 (3.0) | 2 (2.0) | ||
| ALT increased |
— | 3 (3.0) | ||
| ** | Two (2) subjects experienced Serious Adverse Events during this study. Both subjects were randomized to placebo. |
22
Study REC-15-015
In November 2016, we announced positive results from the second of our two pivotal clinical trials, evaluating pain relief over a 24 hour period in a soft tissue, post-operative pain model (abdominoplasty). In the trial, IV meloxicam achieved the primary endpoint of a statistically significant difference in SPID over the first 24 hours, SPID24, compared to placebo. This was a Phase III, randomized, multicenter, multi-dose, double-blind, placebo-controlled study evaluating IV meloxicam in the management of post-operative pain following abdominoplasty surgery. Two hundred nineteen patients who met the eligibility criteria were randomized to receive either IV meloxicam (30 mg) or placebo once daily for three days. Following the beginning of treatment, patients remained under observation for 48 hours at study centers. Patients were followed for 7 days after the initial dose of study medication. There was an oral opioid rescue treatment available to all patients, if required. The primary objective of the trial was to evaluate pain relief over a 24-hour period of IV meloxicam when administered as a bolus injection (over 15-30 seconds).
The primary efficacy endpoint of the trial was SPID24 (0-24), utilizing a W2LOCF analysis method. Secondary efficacy endpoints included use of opioid rescue medication, SPIDs over various time intervals, time to pain relief and PGA of pain control. The IV meloxicam treatment arm demonstrated a statistically significant reduction in SPID24 (p=0.0145) compared to the placebo arm (Figure 2).
Figure 2: SPID24
.
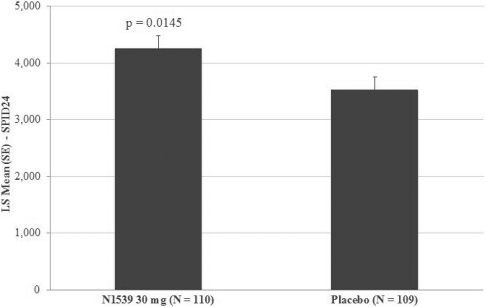
The study also achieved statistical significance for 10 of the secondary endpoints, including statistically significant differences in SPID12 (p=0.0434), time to perceptible pain relief (p=0.0050), subjects with ³30% improvement at 24 hours (p=0.0178), number of times patients required rescue in the first 24 hours after randomization (p=0.0275), as well as number of times rescued from 24 to 48 hours (p=0.0009), and several other pain relief metrics, compared to placebo (Table 3).
23
Table 3: Summary of Secondary Endpoints
| Parameter |
p-value | |||
| SPID12 |
0.0434 | |||
| SPID48 |
0.0040 | |||
| SPID24-48 |
0.0028 | |||
| Number of Subjects Rescued 24-48 Hours |
0.0014 | |||
| Number of Times Rescued 0-24 Hours |
0.0275 | |||
| Number of Times Rescued 24-48 Hours |
0.0009 | |||
| Number of Times Rescued 0-48 Hours |
0.0027 | |||
| Time to Perceptible Pain Relief |
0.0050 | |||
| % Subjects with ³30% Improvement – 24 Hours |
0.0178 | |||
| PGA of Pain Control at 48 hours |
0.0027 | |||
SPID6, Times to Meaningful Pain Relief and First Rescue, Number of Subjects rescued 0-24 and 0-48 hours, % Subjects with ³30 and ³50% Improvement within 6 Hours and ³50% within 24 hours, PGA of Pain Control at 24 hours were not significantly different between treatment groups.
The safety results demonstrated that IV meloxicam was well tolerated with no difference in SAEs related to bleeding for IV meloxicam treated patients versus placebo (1 each). There were two additional SAEs observed in the placebo group. The most common (³2% in the IV meloxicam group) AEs were nausea, headache, vomiting, and dizziness (Table 4). The incidence of these events was lower than those observed in the placebo group. The majority of AEs were mild in nature and one patient in the placebo group discontinued treatment due to an adverse event of post-procedural bleeding. There were no meaningful differences between treatment groups in vital signs, ECGs or clinical lab assessments.
Table 4: Adverse Events reported by ³2% of subjects from any treatment group
| n (%) of Subjects | ||||
| Preferred Term |
N1539 30 mg (N=110) |
Placebo (N=109) | ||
| Subjects with >=1 TEAE |
58 (52.7) | 80 (73.4) | ||
| Nausea |
30 (27.3) | 41 (37.6) | ||
| Headache |
13 (11.8) | 18 (16.5) | ||
| Vomiting |
5 (4.5) | 10 (9.2) | ||
| Dizziness |
4 (3.6) | 10 (9.2) | ||
| ** | Four (4) subjects experienced Serious Adverse Events during this study. Three subjects were randomized to placebo and one to N1539. |
Phase II Clinical Trials
Study REC-15-014
This was a Phase II, randomized, single-center, double-blind, placebo-controlled study evaluating IV meloxicam in the management of post-operative pain following bunionectomy surgery. Fifty-nine patients who met the eligibility criteria were randomized to receive either IV meloxicam (30 mg or 60 mg dosage groups) or placebo once daily for two days. Following the beginning of treatment, patients remained under observation for 48 hours at study centers. Patients were followed for 7 days after the initial dose of study medication. There was an oral opioid rescue treatment available to all patients, if required. The primary objective of the trial was to evaluate the safety of IV meloxicam when administered as a bolus injection (over 15-30 seconds).
The safety results demonstrated that IV meloxicam was well tolerated with no serious adverse events, bleeding events or injection site reactions. The most common AEs were nausea, headache, dizziness,
24
pruritus and vomiting and were comparable to the placebo group. There were no discontinuations due to AEs. The majority of TEAEs were mild in nature and determined by investigators to be “not related” or “possibly related” to study drug. There were no vital signs changes that necessitated treatment. There were no observed changes in the evaluation of ECGs. No clinically meaningful lab changes were observed in the meloxicam treatment groups (Table 5).
Table 5: Adverse Events reported by ³2% of subjects from any treatment group
| Placebo N = 19 n (%) |
IV Meloxicam | |||||||||||
| 30 mg N = 20 n (%) |
60 mg N = 20 n (%) |
|||||||||||
| Nausea |
4 (21.1) | 6 (30.0) | 4 (20.0) | |||||||||
| Headache |
4 (21.1) | 2 (10.0) | 3 (15.0) | |||||||||
| Dizziness |
1 (5.3) | 3 (15.0) | 2 (10.0) | |||||||||
| Pruritus |
0 (0.0) | 1 (5.0) | 2 (10.0) | |||||||||
| Vomiting |
1 (5.3) | 3 (15.0) | 0 (0.0) | |||||||||
| Decreased appetite |
2 (10.5) | 0 (0.0) | 1 (5.0) | |||||||||
| Erythema |
1 (5.3) | 2 (10.0) | 0 (0.0) | |||||||||
| Constipation |
0 (0.0) | 1 (5.0) | 1 (5.0) | |||||||||
| GGT increased |
2 (10.5) | 0 (0.0) | 0 (0.0) | |||||||||
| Muscle spasms |
0 (0.0) | 2 (10.0) | 0 (0.0) | |||||||||
| Somnolence |
0 (0.0) | 1 (5.0) | 1 (5.0) | |||||||||
The primary efficacy endpoint of the trial was SPID48 (0-48), utilizing the last observation carried forward, or LOCF, analysis method. Secondary efficacy endpoints included use of opioid rescue medication, SPIDs over various time intervals, and patient global assessment, or PGA, of pain control. Both the 30 mg and 60 mg IV meloxicam treatment arms demonstrated statistically significant reductions in pain intensity, as measured by SPID48 (p=0.0007 and p=0.0027, respectively) compared to placebo (Figure 3). Although there were observed differences in opioid consumption among the meloxicam dose groups and the placebo group, in general these differences did not meet statistical significance.
Figure 3: SPID48
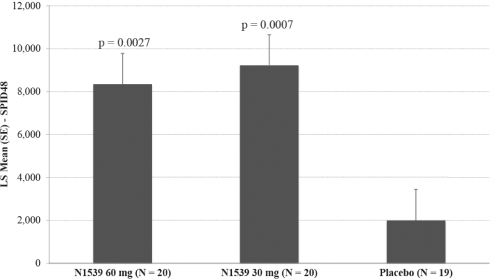
25
Pain intensity was measured at various time points throughout the study. Differences in pain intensity were observed as early as 10 minutes and continued throughout the 48 hour observation period. Overall the 30 mg and 60 mg dose groups performed in a very comparable fashion (Figure 4).
Figure 4: Pain Intensity Differences for Each Time Point
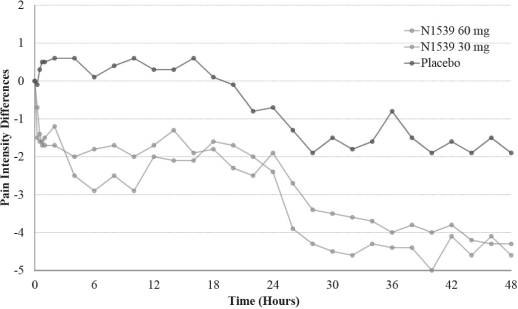
Study N1539-04
This was a Phase II, multicenter, randomized, double-blind, placebo-and active-controlled study in 486 female subjects who underwent open abdominal hysterectomy. Following surgery on post-operative day 1, or Post Op Day 1, subjects received a single dose of either IV placebo, morphine or meloxicam 5 mg, 7.5 mg, 15 mg, 30 mg or 60 mg. Starting at the time of study drug administration and continuing for 24 hours thereafter, subjects had access to rescue medication. During the 24-hour double-blind evaluation period, efficacy measurements of pain intensity and pain relief were made using the 100-mm VAS to assess pain intensity and a 5-point categorical scale (ranging from none to complete) to assess pain relief.
Overall, all active treatment doses produced statistically significant reductions in SPID24 (a co-primary endpoint) compared to placebo (p <0.001), utilizing the LOCF analysis method. In addition, all active treatment doses also produced statistically significant improvement in TOTPAR24 (a co-primary efficacy endpoint) compared to placebo (p <0.001). Statistically significant decreases in pain intensity from baseline were detected as early as 10 minutes post-dose and continued throughout the 24 hour postdose period. In general, the greatest decreases were seen in the 30 mg and 60 mg dose groups followed by the 15 mg group (Figure 5).
26
Figure 5: Pain Intensity Differences at Various Time Points
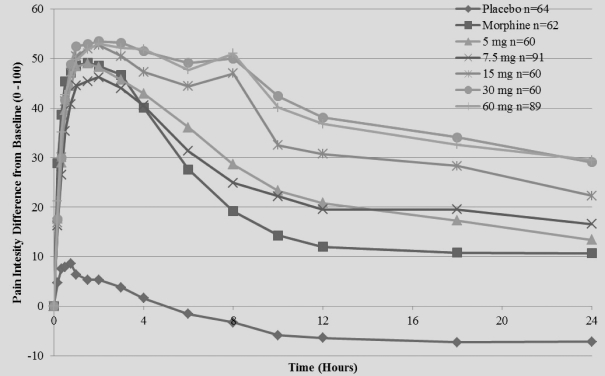
Rescue medication use during the 24-hour double-blind period was reduced by approximately 90% in the meloxicam 30 mg and 60 mg dose groups, and by 86%, 77%, 81%, and 71% in the 15 mg, 7.5 mg, 5 mg, and morphine groups, respectively, compared to placebo. Statistically significant differences were seen between each active group and placebo (p <0.001). The percentage of subjects using rescue medication is presented in Figure 6. The median time to rescue (based on the lower bound of the 95% confidence interval for the 50th percentile) was greatest for meloxicam 30 mg (21.9 hours), followed by 60 mg (20.6 hours), 15 mg (18.3 hours), 5 mg (12.2 hours), 7.5 mg (8.3 hours), morphine (6.6 hours), and placebo (1.1 hours).
27
Figure 6: Percentage of Subjects Using Rescue Medication
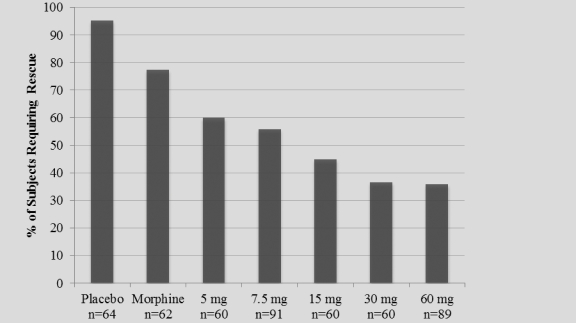
Study medication was well tolerated. A total of five SAEs were reported in the study, and none were assessed as related to treatment. There were no clinically meaningful trends in vital signs, electrocardiograms or laboratory assessments. Adverse event rates were generally low and consistent with this surgical population under study (Table 6).
Table 6: Adverse Events reported by ³3% of subjects from any treatment group
| IV Meloxicam | ||||||||||||||
| Placebo N = 64 n (%) |
Morphine N = 62 n (%) |
5 mg N = 60 n (%) |
7.5 mg N = 91 n (%) |
15 mg N = 60 n (%) |
30 mg N = 60 n (%) |
60 mg N = 89 n (%) | ||||||||
| Anemia |
2 (3.1) | 3 (4.8) | 2 (3.3) | 12 (13.2) | 2 (3.3) | 1 (1.7) | 9 (10.1) | |||||||
| Leukocytosis |
0 (0.0) | 0 (0.0) | 1 (1.7) | 0 (0.0) | 0 (0.0) | 2 (3.3) | 0 (0.0) | |||||||
| Sinus tachycardia |
0 (0.0) | 0 (0.0) | 2 (3.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.1) | |||||||
| Abdominal distension |
2 (3.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||||||
| Constipation |
0 (0.0) | 3 (4.8) | 3 (5.0) | 1 (1.1) | 1 (1.7) | 0 (0.0) | 0 (0.0) | |||||||
| Flatulence |
0 (0.0) | 3 (4.8) | 1 (1.7) | 1 (1.1) | 2 (3.3) | 0 (0.0) | 0 (0.0) | |||||||
| Nausea |
2 (3.1) | 1 (1.6) | 1 (1.7) | 1 (1.1) | 1 (1.7) | 1 (1.7) | 2 (2.2) | |||||||
| Pyrexia |
1 (1.6) | 2 (3.2) | 2 (3.3) | 2 (2.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||||||
| Anemia post-operative |
0 (0.0) | 1 (1.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (3.3) | 0 (0.0) | |||||||
| Hypokalemia |
0 (0.0) | 2 (3.2) | 1 (1.7) | 1 (1.1) | 0 (0.0) | 1 (1.7) | 0 (0.0) | |||||||
| Insomnia |
3 (4.7) | 5 (8.1) | 6 (10.0) | 4 (4.4) | 3 (5.0) | 3 (5.0) | 4 (4.5) | |||||||
| Ketonuria |
5 (7.8) | 6 (9.7) | 4 (6.7) | 9 (9.9) | 9 (15.0) | 6 (10.0) | 9 (10.1) | |||||||
Study N1539-02
This Phase II study was a randomized, double-blind, double-dummy, placebo-controlled, active-controlled, single center study in 230 subjects who underwent third molar extraction surgery. Subjects received a single dose of either IV placebo, oral ibuprofen 400 mg, or IV meloxicam 15 mg, 30 mg or
28
60 mg. Starting at the time of study drug administration and continuing for 24 hours thereafter, subjects were given access to rescue medication for pain not relieved by the study drug. SPID24 was the primary endpoint utilizing the LOCF analysis method for this study.
Overall, the results of this study consistently demonstrated that IV meloxicam produced the greatest reduction in pain, followed by the 30 mg and 15 mg doses, as well as ibuprofen 400 mg. Highly statistically significant differences were seen among the treatments for the primary endpoint, SPID24, as well as in every efficacy analysis.
The onset of action was rapid for the IV meloxicam doses, with statistically significant differences in pain intensity and pain reduction detected among the treatments as early as 10 minutes. For the IV meloxicam doses, analgesia was sustained, with statistically significant differences in pain intensity and pain relief evident through 24 hours postdose.
The use of rescue medication was reduced by 93%, 86%, and 87% by the IV meloxicam 60 mg, 30 mg, and 15 mg doses, respectively, compared to placebo.
Overall, treatment with IV meloxicam was well-tolerated after a single-dose up to 60 mg. There were no SAEs or discontinuations due to an adverse event reported in this study. There were no clinically meaningful trends in vital signs or laboratory assessments. Adverse event rates were generally low and consistent with this surgical population.
Study N1539-05
This study was a Phase II, single-center, randomized, double-blind, placebo- and active-controlled, study conducted in subjects undergoing abdominal laparoscopic surgery. Allowed procedures included biliary tree surgery, common bile duct exploration/surgery, cholecystectomy and inguinal hernia surgery. Subjects received either IV placebo; IV ketorolac every 6 hours; or IV meloxicam 7.5 mg every 12 hours, 15 mg every 12 hours, or 30 mg once daily, for up to 48 hours. Rescue medication was available any time after the initial dose of study drug. The study was expected to enroll 250 subjects. However, the prior sponsor decided to terminate this study for business reasons. A total of 50 subjects had been enrolled prior to the study’s discontinuation. Although a full efficacy analysis was not completed due to the early termination, analysis of the data from the enrolled subjects demonstrated that IV meloxicam 30 mg once daily produced a statistically significant difference compared to placebo for the SPID48.
Overall, study medication was well tolerated. The most frequently reported AEs for all subjects were headache, dry mouth, dysuria, nausea, fatigue and dizziness. There was no apparent trend in occurrence of AEs and treatment group. One SAE was reported by a subject in the ketorolac group. One subject in the IV meloxicam 7.5 mg every 12 hours group discontinued due to maculopapular rash.
Pharmacokinetic Studies
Pharmacokinetic studies have examined single and multiple doses of IV meloxicam. In general terms, IV administration resulted in peak plasma concentrations immediately follow dosing. When compared to oral Mobic, IV meloxicam had similar areas under the plasma drug concentration-time curve and half-lives for doses of 15 mg, 30 mg and 60 mg.
Recro Gainesville
Through our subsidiary, Recro Gainesville, we leverage our formulation and development expertise to develop and manufacture pharmaceutical products using our proprietary delivery technologies for commercial partners who commercialize or plan to commercialize these products. Our manufacturing and development capabilities include formulation through process development, scale-up and full-scale
29
commercial manufacturing and specialized capabilities for the development and manufacturing of controlled substances. In a typical collaboration, we license certain intellectual property to our commercial partners and work with our commercial partners to develop product candidates, or new formulations of existing product candidates. We also typically exclusively manufacture and supply clinical and commercial supplies of these product candidates. These collaborations result in revenue streams including from royalties, profit sharing, research and development and manufacturing, which support continued operations for Recro Gainesville as well as our research and development of proprietary product candidates.
The table below details the key products developed and/or manufactured with our commercial partners:
| Product |
Indication | Technology | Territory | Revenue Source | Commercial Partner | |||||
| Ritalin LA® | Attention Deficit Hyperactivity Disorder |
OCR (SODAS) | Worldwide | Royalty Manufacturing |
Novartis Pharma AG | |||||
| Focalin XR® | Attention Deficit Hyperactivity Disorder |
OCR (SODAS) | Worldwide, except Canada |
Royalty Manufacturing |
Novartis Pharma AG | |||||
| Verelan PM® | Hypertension | OCR (SODAS) | United States | Royalty Manufacturing |
Lannett Company, Inc. | |||||
| Verapamil (generic) | Hypertension | OCR (SODAS) | United States | Profit Sharing Manufacturing |
Teva Pharmaceutical Industries Ltd. | |||||
| Zohydro ER® | Severe Pain | OCR (SODAS) | United States | Royalty Manufacturing |
Pernix Therapeutics, Inc. | |||||
| Canada | Royalty Manufacturing |
Paladin Labs, Inc. |
In addition to these key products, we also develop and manufacture other development stage products. The manufacture of these products for clinical trials and commercial use is subject to cGMPs and other regulatory agency regulations. We own and operate a 97,000 square foot, DEA-licensed manufacturing facility in Gainesville, Georgia, which has been inspected by U.S., EU, Turkish and Brazilian regulatory authorities for compliance with required cGMP standards for continued commercial manufacturing.
With each product, we either purchase active drug product from third parties or receive it from our commercial partners to formulate product using our technologies. Although some materials for our products are currently available from a single source or a limited number of qualified sources, we attempt to acquire an adequate inventory of such materials, establish alternative sources and/or negotiate long-term supply arrangements. We do not currently have any significant issues finding suppliers. However, there is no certainty that we will be able to obtain long-term supplies of our manufacturing materials in the future.
Permits and Regulatory Approvals
We hold various licenses for our Gainesville manufacturing activities. The primary licenses held are FDA Registrations of Drug Establishments and DEA Controlled Substance Registration. Due to certain U.S. state law requirements, we also hold certain state licenses for distribution activities throughout certain states. We also hold cGMP certifications for EU importation of products made in Gainvesville for sale in the EU.
We do not generally act as the product authorization holder for products that have been developed on behalf of a commercial partner. In such cases, our commercial partner typically holds the relevant authorization from the FDA or other national regulator, and we support this authorization by furnishing
30
a copy of the Drug Master File, or the chemistry, and manufacturing and related data to the relevant regulator or sponsor to provide adequate manufacturing support in respect of the product. We generally update this information annually with the relevant regulator.
Customer Agreements
We are party to agreements with each of our commercial partners governing the development, formulation and/or supply services were provide, as well as any applicable intellectual property licenses. Each commercial partner generally remains responsible for distributing, marketing and promoting their respective products. These collaborations result in revenue streams including royalties, profit sharing, research and development and manufacturing, which support continued operations for Gainesville as well as our research and development of proprietary product candidates. We are dependent on a small number of commercial partners, with our four largest customers (Teva Pharmaceutical Industries, Inc., Novartis AG, Pernix Therapeutics, Inc. and Lannett Company, Inc.) having generated 96% of our revenues for the nine months ended September 30, 2016, of which one customer generated 43% of our revenue, and another customer generated 38% of our revenue under two separate customer agreements.
During the nine months ended September 30, 2016 and year ended December 31, 2015, revenue under one of our customer agreements, with Watson Laboratories, Inc., a subsidiary of Teva Pharmaceutical Industries Ltd., or Teva, represented 43% and 33% of our revenues, respectively. Pursuant to the amended and restated license and supply agreement, or the License and Supply Agreement, between us and Watson Laboratories, Inc., we exclusively manufacture generic Verapamil for Teva. We receive a percentage profit share from Teva on all U.S. sales of Verapamil and are compensated for manufacturing the product at cost (or, where product is supplied in finished form, at manufacturing cost plus a mark-up). Under the License and Supply Agreement, we also license certain intellectual property to Teva and maintain the regulatory approval that is necessary to enable Teva to distribute Verapamil in the United States. Teva is responsible for distributing, marketing and promoting Verapamil in the United States. The License and Supply Agreement also contains certain restrictions in respect of manufacturing and selling competing products, although we are permitted to sell a branded version of Verapamil through a third party under the trade name Verelan®. The License and Supply Agreement renews for one-year terms annually unless terminated by either party by providing notice in advance of the renewal date.
Our Pipeline Candidates
Dex
Dex is a selective alpha-2 adrenergic agonist that has demonstrated sedative, analgesic and anxiolytic properties. Hospira currently markets IV Dex as a sedative trademarked Precedex® in the United States, and Orion markets IV Dex as an ICU sedative in the European Union, trademarked as Dexdor®. Dex has an extensive history of safe intravenous use. We have formulated Dex-IN, a proprietary intranasal formulation of Dex, at a significantly lower dose (perhaps as low as 1/10th) than the currently recommended IV dosage levels. Based upon our lower dose, we have seen minimal sedation to date in our clinical trials while still demonstrating an analgesic effect.
We initially studied Dex-IN for the treatment of post-operative pain. Based on feedback from the FDA regarding Dex-IN’s benefit-risk profile, specifically its efficacy and blood pressure effects, which was demonstrated in post-operative pain, and the subsequent requirements for a post-operative pain clinical program, we determined not to pursue Dex-IN in post-operative pain due to time, cost and associated risk. We are exploring the possibility of evaluating Dex-IN in peri-procedural pain. If approved, Dex-IN would also be the first and only approved peri-procedural pain drug in its class of drugs.
We also have a sublingual formulation of Dex, Dex-SL, which may be appropriate for use in treating chronic pain.
31
Fado
We also have another selective alpha-2 agonist product candidate in our pipeline, Fado. Fado is similar to Dex and different from clonidine in that it is a full agonist of all subtypes of alpha-2 adrenoreceptor. Unlike Dex, Fado does not cross the blood/brain barrier, and this accounts for the targeting of Fado use for either intrathecal administration for pain or anesthesia, or potentially for topical use to treat pain associated with regional nerve pain from underlying nerve damage, also called “neuropathies.” Various preclinical models of pain have been employed and have demonstrated Fado’s potential as an analgesic, including its potential for use in neuropathies and post-operative pain. In Orion sponsored studies, Fado appeared to delay the onset of pain while doses of Fado greater than 120 mcg also appeared to suppress pain. In addition, Fado was well tolerated by subjects.
Intellectual Property
Injectable Meloxicam
We own patents and patent applications for injectable meloxicam, that cover compositions, including compositions produced using NanoCrystal® technology, method of making and method of treating. These issued patents expire in 2022 in the United States. We also in-license from Alkermes, on a perpetual, royalty-free basis, composition and methods of making patent and patent applications (specifically directed to the prevention of flake like substances) which expire in 2030.
Recro Gainesville
We also own various controlled release formulation patents, including patents in the United States, Canada, and Europe, related to our proprietary delivery technologies that we utilize in our drug development, formulation and manufacturing business through Recro Gainesville. These patents are scheduled to expire between 2019 and 2026. We own patents and patent applications in the United States and Canada directed to the composition of, manufacturing of, and formulating of Zohydro ER®. We license our U.S. patents and patent applications to our commercial partner, Pernix Therapeutics Holdings, Inc., or Pernix, in the United States. We also own Canadian patents and patent applications relating to the same technology, which we license to our commercial partner, Paladin Labs Inc., in Canada. The patent protection for Zohydro ER® provides for protection of Zohydro ER® through 2034, subject to any extensions or disclaimers.
Our Pipeline Candidates
We hold patent applications directed to the analgesia indication, formulations and intranasal and transmucosal methods of use of Dex, and we are progressing through the patent application process globally, including the United States. Several patent applications have issued as patents outside the United States for transmucosal methods, and the resulting patent protection will last into 2030, subject to any disclaimers or extensions. If the intranasal patent applications are issued as patents, the resulting patent protection will last into 2032, subject to any disclaimers or extensions. Also for Fado, we have a pro-drug patent that expires in 2025.
We are party to an exclusive license with Orion for the development and commercialization of Dex for use in the treatment of pain in humans in any dosage form for transdermal, transmucosal (including sublingual and intranasal), topical, enteral or pulmonary (inhalational) delivery, but specifically excluding delivery vehicles for administration by injection or infusion, worldwide, except for Europe, Turkey, and the CIS (currently includes Armenia, Azerbaijan, Belarus, Georgia, Kazakhstan, Kyrgyzstan, Moldova, Russia, Tajikistan, Turkmenistan, Ukraine and Uzbekistan), referred to herein as the Territory. We have the right to sublicense the rights under such license at any time. We are required to pay Orion lump sum payments on the achievement of certain developmental milestones and upon the achievement of certain commercial milestones, as well as a royalty on net sales during the term, which varies from
32
10% to 20% depending on annual sales levels. We will pay milestone payments to Orion of up to €20.5 million ($23.0 million as of September 30, 2016) after regulatory approval of Dex dosage forms and upon achieving certain sales milestones. Through September 30, 2016, no such milestones have been achieved. The initial term of this license is 15 years from the first commercial sale in the Territory, with automatic two year extensions, unless either party provides written notice of termination.
We are also party to an exclusive license agreement with Orion for the development and commercialization of Fado for use as a human therapeutic, in any dosage form in the Territory. We have the right to sublicense the rights under such license at any time. In consideration for this license, we paid Orion an upfront payment and are required to pay certain lump-sum amounts on completion of certain development milestones, as well as on achievement of certain commercial milestones. We will pay milestone payments to Orion of up to €12.2 million ($13.7 million as of September 30, 2016), based on regulatory filings and approval and on commercialized net sales levels. We will also pay Orion royalty payments on net sales of Fado ranging from 10% to 15%. Through September 30, 2016, no such milestones have been achieved. The initial term of this license is 15 years from the first commercial sale in the Territory, with automatic three year extensions, unless either party provides written notice of termination at least six months prior to expiration or unless otherwise terminated pursuant to the terms of the license agreement.
Intellectual Property Protection Strategy
We intend to rely on a combination of patents and trade secrets, as well as confidentiality agreements and license agreements, to protect our product candidates. Our patent strategy is designed to facilitate commercialization of our current product candidates and future product candidates, as well as create barriers to entry for third parties. One focus of our claim strategy is on formulation claims and method of treatment claims.
We are seeking patent protection in the United States and internationally for our product candidates. Our policy is to pursue, maintain and defend patent rights and to protect the technology, inventions and improvements that are commercially important to the development of our business. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents granted to us in the future will be commercially useful in protecting our technology. We also intend to rely on trade secrets to protect our product candidates. Our commercial success also depends in part on our non-infringement of the patents or proprietary rights of third parties.
Our success will depend significantly on our ability to:
| • | obtain and maintain patent and other proprietary protection for our product candidates; |
| • | defend our patents; |
| • | develop trade secrets as needed and preserve the confidentiality of our trade secrets; and |
| • | operate our business without infringing the patents and proprietary rights of third parties. |
We have taken steps to build and will continue to build proprietary positions for our product candidates and related technology in the United States and abroad. We note that the patent laws of foreign countries differ from those in the United States, and the degree of protection afforded by foreign patents may be different from the protection offered by United States patents.
Sales and Marketing
Our current intent is to develop and commercialize our product candidates in the United States while out-licensing development and commercialization rights for other territories outside the United States for
33
which we own the territorial rights. We believe the initial target audience for our product candidates will be specialty physicians, including surgeons, anesthesiologists and pain specialists. Our management team has experience building and launching therapeutics to specialty physicians. As this target audience is smaller than general practitioners, we believe we have the capabilities to build a sales and marketing infrastructure and effectively market our product candidates upon commercial approval. While our stated intention is to develop and commercialize our product candidates, we will evaluate potential strategic collaborations that could accelerate or enhance our development and, upon approval, commercial success of our product candidates.
We are currently preparing for a potential U.S. commercial launch of IV meloxicam, if approved, and we plan to establish sales, marketing and reimbursement functions to commercialize IV meloxicam in the United States.
Manufacturing and Supply of our Product Candidates
We currently rely on contract manufacturers to produce drug product for our clinical studies under cGMPs, with oversight by our internal managers. We plan to continue to rely on contract manufacturers to manufacture development quantities of our product candidates, as well as commercial quantities of our product candidates, if and when approved for marketing by the FDA. We currently rely on a single manufacturer for the clinical supplies of our drug product for each of our product candidates and do not currently have agreements in place for redundant supply or a second source for any of our product candidates. We have identified other drug product manufacturers that could satisfy our clinical study requirements, but this would require significant expense and could produce a significant delay in setting up the facility and moving equipment. Additionally, should a supplier or a manufacturer on whom we rely to produce a product candidate provide us with a faulty product or a product that is later recalled, we would likely experience significant delays and additional costs.
Injectable Meloxicam
Pursuant to a Development, Manufacturing and Supply Agreement, or Supply Agreement, Alkermes (through a subsidiary), will provide (i) clinical and commercial bulk supplies of IV meloxicam formulation, and (ii) development services with respect to the Chemistry, Manufacturing and Controls section of the NDA for IV meloxicam. Pursuant to the Supply Agreement, Alkermes will supply us with such quantities of bulk IV meloxicam formulation as shall be reasonably required for the completion of clinical trials of IV meloxicam, subject to a maximum of eight clinical batches in any twelve-month period, unless otherwise agreed by the parties. During the term of the Supply Agreement, we will purchase our clinical and commercial supplies of bulk IV meloxicam formulation exclusively from Alkermes. Sterile fill-finish of Meloxicam will be completed by a third party fill-finish facility. If the first commercial sale of meloxicam occurs on or prior to December 31, 2020, the Supply Agreement will have an initial term expiring ten years following the date of such first commercial sale. The Supply Agreement will then automatically renew for successive one-year terms unless terminated by either party upon written notice at least 180 days prior to the expiration of the applicable term. If the first commercial sale of meloxicam has not occurred by December 31, 2020, the Supply Agreement will expire on that date.
The Supply Agreement may be terminated earlier (i) by us upon 180 days’ written notice following the date of first generic entry; (ii) by either party upon twelve months’ written notice following the first anniversary of the approval of the NDA for meloxicam; (iii) by either party upon written notice to the other party in the event of uncured material breach of the other party; and (iv) by Alkermes upon written notice in certain events of uncured non-payment.
Dex and Fado
We are party to an API supply agreement with Orion, whereby Orion provides us with API for the development and commercialization of the Dex and Fado product candidates. Prior to obtaining
34
regulatory approval, subject to advance notice to Orion, Orion will provide API without charge for agreed upon amounts. Any amounts ordered by us that are greater than the planned supply will be charged at 50% of the supply price for commercial product. The initial term of the agreement is the later of 15 years from the first commercial sale and 15 years after the effective date of the agreement, and in each case, will be automatically extended for one or more periods of two years unless terminated. After the initial term, the agreement may be terminated upon six months’ notice to the other party.
The single unit dose intranasal sprayer for Dex is manufactured by a supplier of proprietary components and devices, and equipment is leased from the device supplier for filling at a contract manufacturer. It is possible that we will continue with this arrangement through clinical development, evaluate the option of entering a manufacturing agreement with the device originator or evaluate alternative devices prior to commercialization. Suppliers of components, subassemblies and other materials are located in Europe, Asia and the United States.
Competition
The pharmaceutical and biotechnology industries are intensely competitive and subject to rapid and significant technological change. Our current and future competitors include pharmaceutical, biotechnology and specialty pharmaceutical companies. Many of our competitors have greater financial and other resources than we have, such as more commercial resources, larger research and development staffs and more extensive marketing and manufacturing organizations. As a result, these companies may obtain marketing approval more rapidly than we are able to obtain and may be more effective in selling and marketing their products. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies.
Our competitors may succeed in developing, acquiring or licensing technologies and drug products that are more effective or less costly than our product candidates or any other products that we may develop which could render our products obsolete and noncompetitive. We expect any products that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, convenience of administration and delivery, price and the availability of reimbursement from government and other third-party payors. We also expect to face competition in our efforts to identify appropriate collaborators or partners to help commercialize our product candidates in our target commercial markets.
In the post-operative pain relief setting, we believe patients are prescribed injectable acetaminophen, NSAIDs, sodium channel blockers and opioids, depending on the severity of pain. Specifically, acetaminophen, NSAIDs and sodium channel blockers, we believe, are prescribed for mild to moderate pain relief, whereas we believe opioids are prescribed for moderate to severe pain relief. While we will compete with all of these compounds in the post-operative pain setting, we believe injectable meloxicam will be prescribed for moderate to severe pain, competing with opioids and other non-opioid pain treatments. There are a number of pharmaceutical companies that currently market therapeutics in the pain relief area, including Johnson & Johnson, Purdue Pharma, L.P., Endo Pharmaceuticals, Inc., Mallinckrodt plc and Pacira Pharmaceuticals, Inc. Purdue and Endo are the primary competitors in the manufacture, marketing and commercialization of opioid therapeutics. Mallinckrodt commercializes an injectable formulation of acetaminophen. Pacira commercializes an intraoperative formulation of bupivacaine, a sodium channel blocker. Additionally, companies such as Adynxx, Inc., AcelRx Pharmaceuticals, Inc., Durect Corporation, Heron Therapeutics, Inc., Pacira, Trevena, Inc. and Cara Therapeutics, Inc. are currently developing post-operative pain therapeutics that could compete with us in the future.
With our development, formulation and manufacturing business, we compete with contract pharmaceutical formulation and manufacturing companies such as Catalent, Inc., Patheon Holdings
35
Coôperatief U.A., Adare Pharmaceuticals, Inc., Metrics, Inc., a subsidiary of Mayne Pharma Group Limited, and other packaging and manufacture-related service providers.
Government Regulation
Governmental authorities in the United States at the federal, state and local level, and other in countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, marketing, export and import of products such as those we are developing. Our product candidates, including our formulations of injectable meloxicam, Dex and Fado, must be approved by the FDA before they may legally be marketed in the United States.
U.S. Drug Development Process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations. The process of obtaining regulatory approvals and ensuring compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process, or after approval, may subject an applicant to administrative enforcement or judicial sanctions. This enforcement could include, without limitation, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, corrective actions, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| • | completion of preclinical laboratory tests, animal studies and formulation studies, some of which must be conducted according to Good Laboratory Practices regulations; |
| • | submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| • | performance of adequate and well-controlled human clinical trials according to the FDA’s current Good Clinical Practices, or cGCPs, to establish the safety and efficacy of the proposed drug for its intended use; |
| • | submission to the FDA of an NDA for a new drug; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities identified in the NDA; and |
| • | FDA review and approval of the NDA. |
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all.
Once a pharmaceutical product candidate is identified for development, it enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity, formulation and stability, as well as animal studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, to the FDA as part of the IND. The sponsor must also include a protocol detailing, among other things, the objectives of the initial clinical trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated if the initial clinical trial lends itself to an efficacy evaluation. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days
36
after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during trials due to safety concerns regarding the product candidate or non-compliance with applicable requirements.
All clinical trials of a product candidate must be conducted under the supervision of one or more qualified investigators, in accordance with cGCP regulations. These regulations include the requirement that all research subjects provide informed consent. Further, an institutional review board, or IRB, must review and approve the plan for any clinical trial before it commences at any institution. The IRB’s role is to protect the rights and welfare of human subjects involved in clinical studies by evaluating, among other things, the potential risks and benefits to subjects, processes for obtaining informed consent, monitoring of data to ensure subject safety, and provisions to protect the subjects’ privacy. The IRB approves the information regarding the clinical trial and the consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
Once an IND is in effect, each new clinical protocol, and any amendments to the protocol, must be submitted to the IND for FDA review and to the IRBs for approval. Protocols detail, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| • | Phase I. The product is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing may be conducted in patients. |
| • | Phase II. Phase II trials involve investigations in a limited patient population to identify possible AEs and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted indications and to determine dosage tolerance and optimal dosage and schedule. |
| • | Phase III. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for regulatory approval and product labeling. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA, and safety reports must be submitted to the FDA and the investigators for serious and unexpected side effects. Phase I, Phase II and Phase III testing may not be completed successfully within any specified period, if at all. Results from earlier trials are not necessarily predictive of results from later trials. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the
37
identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Review and Approval Processes
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the drug, proposed labeling and other relevant information, are submitted to the FDA as part of an NDA for a new drug, requesting approval to market the product.
The submission of an NDA is subject to the payment of a substantial user fee for a human drug application. A waiver of such fee may be obtained under certain limited circumstances. For example, an applicant is eligible for waiver of the application fee if the applicant is a small business submitting its first human drug application and does not have another product approved under a human drug application and introduced and delivered for introduction into interstate commerce.
In addition, under the Pediatric Research Equity Act of 2003, or PREA, an NDA or supplement to an NDA for a new indication, dosage form, dosing regimen, route of administration, or active ingredient, must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may waive or defer pediatric studies under certain circumstances.
Section 505(b)(2) New Drug Applications. As an alternate path to FDA approval, particularly for modifications to drug products previously approved by the FDA, an applicant may submit an NDA under Section 505(b)(2) of the FDCA (“Section 505(b)(2) NDA”). Section 505(b)(2) was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments, and it permits approval of applications other than those for duplicate products and permits reliance for such approvals on literature or on the FDA’s findings of safety and effectiveness of an approved drug product. A Section 505(b)(2) NDA is an application where at least some of the information required for approval comes from clinical trials not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The FDA requires submission of information needed to support any changes relative to a previously approved drug, known as the reference product, such as published data or new studies conducted by the applicant, including bioavailability or bioequivalence studies, or clinical trials demonstrating safety and effectiveness. The FDA may then approve the Section 505(b)(2) NDA for all or some of the labeled indications for which the reference product has been approved, as well as for any new indication sought by the applicant, unless such indications or uses are protected by patent or exclusivity provisions covering the reference product. To the extent that a Section 505(b)(2) NDA relies on clinical trials conducted for a previously approved drug product or the FDA’s prior findings of safety and effectiveness for a previously approved drug product, the Section 505(b)(2) applicant must submit patent certifications in its application with respect to any patents for the reference product that are listed in the FDA’s publication, Approved Drug Products with Therapeutic Equivalence Evaluations, commonly referred to as the Orange Book. Specifically, the applicant must certify for each listed patent that, in relevant part, (1) the required patent information has not been filed; (2) the listed patent has expired; (3) the listed patent has not expired, but will expire on a particular date and approval is not sought until after patent expiration; or (4) the listed patent is invalid, unenforceable or will not be infringed by the proposed new product. A certification that the new product will not infringe the previously approved product’s listed patent or that such patent is invalid or unenforceable is known as a Paragraph IV certification. If the applicant does not challenge one or more listed patents through a Paragraph IV certification, the FDA will not approve the Section 505(b)(2) NDA application until all the listed patents claiming the referenced product have expired.
38
Further, the FDA will also not approve a Section 505(b)(2) NDA application until any non-patent exclusivity, such as, for example, five-year exclusivity for obtaining approval of a new chemical entity, three-year exclusivity for an approval based on new clinical trials, or pediatric exclusivity, listed in the Orange Book for the reference product, has expired.
If the Section 505(b)(2) NDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the owner of the reference product and relevant patent holders within 20 days after the Section 505(b)(2) NDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement suit against the Section 505(b)(2) applicant. Under the FDCA, the filing of a patent infringement lawsuit within 45 days of receipt of the notification regarding a Paragraph IV certification automatically prevents the FDA from approving the Section 505(b)(2) NDA for 30 months, beginning on the date the patent holder receives notice, or until the patent expires or a court deems the patent unenforceable, invalid or not infringed, whichever is earlier. Even if a patent infringement claim is not brought within the 45-day period, a patent infringement claim may be brought under traditional patent law, but it does not invoke the 30-month stay. Moreover, in cases where a Section 505(b)(2) application containing a Paragraph IV certification is submitted after the fourth year of a previously approved drug’s five year exclusivity period, and the patent holder brings suit within 45 days of notice of certification, the 30-month period is automatically extended to prevent approval of the Section 505(b)(2) application until the date that is seven and one-half years after approval of the previously approved reference product. The court also has the ability to shorten or lengthen either the 30-month or the seven and one-half year period if either party is found not to be reasonably cooperating in expediting the litigation. Thus, the Section 505(b)(2) applicant may invest a significant amount of time and expense in the development of its product only to be subject to significant delay and patent litigation before its product may be commercialized. Alternatively, if the NDA applicant or relevant patent holder does not file a patent infringement lawsuit within the specified 45 day period, the FDA may approve the Section 505(b)(2) application at any time, assuming the application is otherwise approvable.
Notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over the last few years, some pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2). If the FDA changes its interpretation of Section 505(b)(2), or if the FDA’s interpretation is successfully challenged in court, this could delay or even prevent the FDA from approving any Section 505(b)(2) NDA that we submit.
FDA Review of New Drug Applications. The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. If the FDA does not find an NDA to be sufficiently complete for filing, it may request additional information rather than accepting the NDA for filing. In this event, the sponsor must resubmit the NDA with the additional information. The re-submitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether clinical data demonstrates that a product is safe and effective for its intended use and whether its manufacturing process can assure the product’s identity, strength, quality and purity. Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The FDA may refer the NDA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. An advisory committee is a panel of independent experts who provide advice and recommendations when requested by the FDA. The FDA is not bound by the recommendation of an advisory committee.
The approval process is lengthy and difficult, and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and
39
information. Even if such data and information are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA in its present form. The complete response letter usually describes all of the specific deficiencies that the FDA identified in the NDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, withdraw the application or request an opportunity for a hearing.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages, or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling, and the agency also may require a Risk Evaluation and Mitigation Strategy, or REMS, if it determines that a REMS is necessary to assure that the benefits of a drug outweigh its risks. In addition, the FDA may require Phase IV testing, which involves clinical trials designed to further assess a drug’s safety and effectiveness after NDA approval, and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specific circumstances of FDA marketing approval of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. Subject to certain limitations, the patent term restoration period is generally equal to one-half of the time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. However, each phase of the regulatory review period may be reduced by any time that the FDA finds the applicant did act not act with due diligence. Only one patent applicable to an approved drug is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent and within sixty days of approval of the drug. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for patents that issue from some of our currently owned or licensed patents or patent applications to add patent life beyond their current expiration dates, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA.
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to new drug applications for products containing chemical entities never previously approved by the FDA alone or in combination. A new chemical entity means a drug that contains no active moiety that has been approved by the FDA in any application submitted under Section 505(b) of the FDCA. An active moiety is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a Section 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. This exclusivity provision does not prevent the submission or approval of another full Section 505(b)(1) NDA, but such an NDA applicant would be required to conduct its own preclinical and adequate, well-controlled clinical trials to demonstrate safety and effectiveness. The FDCA also provides three years of marketing exclusivity for an NDA, Section 505(b)(2) NDA or supplement to an existing NDA if new
40
clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application. Such clinical trials may, for example, support new indications, dosages, routes of administration or strengths of an existing drug, or for a new use. This exclusivity, which is sometimes referred to as clinical investigation exclusivity, prevents the FDA from approving an application under a Section 505(b)(2) NDA or an ANDA for the same conditions of use associated with the new clinical investigations before the expiration of three years from the date of approval. Such three-year exclusivity, however, would not prevent the approval of another application if the applicant submits a Section 505(b)(1) NDA and has conducted its own adequate, well-controlled clinical trials demonstrating safety and efficacy, nor would it prevent approval of an ANDA or a Section 505(b)(2) NDA product that did not incorporate the exclusivity-protected aspects of the approved drug product.
Pediatric exclusivity is another type of exclusivity in the United States. Pediatric exclusivity, if granted, provides an additional six months of exclusivity to any existing exclusivity (e.g., three- or five-year exclusivity) or patent protection for a drug. This six month exclusivity, which runs from the end of other exclusivity or patent protection, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
Post-Approval Requirements
Any drugs for which we receive FDA approval will be subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, and complying with FDA promotion and advertising requirements.
The FDA strictly regulates marketing, labeling, advertising, and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other government agencies enforce the laws and regulations prohibiting the false or misleading promotion of drugs.
Further, manufacturers of drugs must continue to comply with cGMP requirements, which are extensive and require considerable time, resources and ongoing investment to ensure compliance. In addition, changes to the manufacturing process may require prior FDA approval before being implemented, and other types of changes to the approved product, such as adding new indications and additional labeling claims, are subject to further FDA review and approval. Drug manufacturers and other entities involved in the manufacturing and distribution of approved drugs are required to list their products and to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage and shipment of the drug. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory standards and test each product batch or lot prior to its release. We rely, and expect to continue to rely, on third parties for the production of clinical quantities of our product candidates. Future FDA and state inspections may identify compliance issues at our site or at the facilities of our contract manufacturers that may disrupt production or distribution or may require substantial resources to correct.
The FDA may withdraw a product approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, untitled and warning letters, holds on clinical trials, product recalls or seizures, product detention or refusal to permit the import or export of
41
products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, consent decrees, injunctions or the imposition of civil or criminal penalties.
From time to time, legislation is drafted, introduced and passed in the U.S. Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition to new legislation, the FDA regulations and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our product candidates. It is impossible to predict whether further legislative or FDA regulation or policy changes will be enacted or implemented and what the impact of such changes, if any, may be.
The U.S. Drug Enforcement Administration
Certain products that we manufacture are regulated as a “controlled substance” as defined in the Controlled Substances Act of 1970, or CSA, which establishes registration, security, recordkeeping, reporting, storage, distribution and other requirements administered and enforced by the DEA. The DEA is concerned with the control and handling of controlled substances, and with the equipment and raw materials used in their manufacture and packaging, in order to prevent loss and diversion into illicit channels of commerce.
The DEA regulates controlled substances by controlling them in five schedules. Schedule I and II controlled substances have a high potential for abuse, whereas Schedule III-V controlled substances have relatively decreasing potential for abuse. Therefore, DEA imposes more stringent controls on Schedule I and II substances than Schedule III-V substances, including stricter security controls, quotas, and increased recordkeeping and reporting requirements. Certain of our products are regulated as Schedule II controlled substances. The DEA establishes annually an aggregate quota for how much certain controlled substances that we manufacture may be produced in total in the United States, based on the DEA’s estimate of the quantity needed to meet legitimate scientific and medicinal needs. This limited aggregate amount that the DEA allows to be produced in the United States each year is allocated among individual companies, who must submit applications annually to the DEA for individual production and procurement quotas. We must receive an annual quota from the DEA in order to produce any Schedule II substance. The DEA may adjust aggregate production quotas and individual production and procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments. Annual registration is required for any facility that manufactures, distributes, dispenses, imports or exports any controlled substance. The registration is specific to the particular location, activity and controlled substance schedule.
The DEA requires facilities that manufacture controlled substances to adhere to certain security requirements. Security requirements vary by controlled substance schedule, with the most stringent requirements applying to Schedule I and Schedule II substances. Required security measures include background checks on employees and physical control of inventory through measures such as cages, surveillance cameras and inventory reconciliations. Records must be maintained for the handling of all controlled substances and periodic reports must be made to the DEA, for example, distribution, acquisition, and inventory reports for Schedule I and II controlled substances, Schedule III substances that are narcotics and other designated substances. Reports must also be made for thefts or losses of any controlled substance and suspicious orders. In addition, special authorization and notification requirements apply to imports and exports.
Failure to maintain compliance with applicable requirements, particularly where noncompliance results in loss or diversion, can result in enforcement action that could have a material adverse effect on our business, results of operations and financial condition. The DEA may seek civil penalties, refuse to renew necessary registrations or initiate proceedings to revoke those registrations, or take other enforcement action. In certain circumstances, violations could result in criminal prosecution.
42
There is a risk that DEA regulations may interfere with the manufacture and supply of the drugs sold commercially, and thus with our ability to produce products in the volume needed to meet commercial demand.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our product candidates to the extent we choose to clinically evaluate or sell any products outside of the United States. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. As in the United States, post-approval regulatory requirements, such as those regarding product manufacture, marketing or distribution, would apply to any product that is approved outside the United States.
Third Party Payor Coverage and Reimbursement
In both the United States and foreign markets, our ability to commercialize our product candidates successfully, and to attract commercialization partners for our product candidates, depends in significant part on the availability of adequate financial coverage and reimbursement from third party payors, including, in the United States, governmental payors such as the Medicare and Medicaid programs, managed care organizations, and private health insurers. Medicare is a federally funded program managed by the Centers for Medicare and Medicaid Services, or CMS, through local Medicare Administrative Contractors that administer coverage and reimbursement for certain healthcare items and services furnished to the elderly and disabled. Medicaid is an insurance program for certain categories of patients whose income and assets fall below state defined levels and who are otherwise uninsured that is both federally and state funded and managed by each state. The federal government sets general guidelines for Medicaid and each state creates specific regulations that govern its individual program. Each government or private payor has its own process and standards for determining whether it will cover and reimburse a procedure or particular product and how much it will pay for that procedure or product. Private payors often rely on the lead of the governmental payors in rendering coverage and reimbursement determinations. Therefore, achieving favorable CMS coverage and reimbursement is usually a significant gating issue for successful introduction of a new product. The competitive position of some of our products will depend, in part, upon the extent of coverage and adequate reimbursement for such products and for the procedures in which such products are used. Prices at which we or our customers seek reimbursement for our product candidates can be subject to challenge, reduction or denial by the government and other payors.
The U.S. Congress and state legislatures may, from time to time, propose and adopt initiatives aimed at cost containment, which could impact our ability to sell our product candidates profitably. For example, in March 2010, President Obama signed into law the Patient Protection and Affordable Care Act and the associated reconciliation bill, which we refer to collectively as the Health Care Reform Law, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Effective October 1, 2010, the Health Care Reform Law revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states once the provision is effective. Further, beginning in 2011, the law imposed a significant annual fee on companies that manufacture or import branded prescription drug products. Substantial provisions affecting compliance have also been enacted. In the coming years, additional changes could be made to the Health Care Reform Law and to governmental healthcare programs that could significantly impact the success of our product candidates.
43
The cost of pharmaceuticals continues to generate substantial governmental and third party payor interest. We expect that the pharmaceutical industry will experience pricing pressures due to the trend toward managed healthcare, the increasing influence of managed care organizations and additional legislative proposals. Our results of operations could be adversely affected by current and future healthcare reforms.
Some third party payors also require pre-approval of coverage for new or innovative devices or drug therapies before they will reimburse healthcare providers that use such therapies. While we cannot predict whether any proposed cost-containment measures will be adopted or otherwise implemented in the future, the announcement or adoption of these proposals could have a material adverse effect on our ability to obtain adequate prices for our product candidates and operate profitably.
Other Healthcare Laws and Compliance Requirements
If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our activities may become subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute, the federal civil False Claims Act, and physician sunshine laws and regulations. These laws and regulations are interpreted and enforced by various federal, state and local authorities including CMS, the Office of Inspector General for the U.S. Department of Health and Human Services, or OIG, the U.S. Department of Justice, individual U.S. Attorney offices within the Department of Justice, and state and local governments. These laws include:
| • | the federal healthcare programs Anti-Kickback Statute, or AKS, which prohibits individuals and entities from, among other things, knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order, or recommendation of an item or service for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value. The government can establish a violation of the AKS without proving that the individual or entity had actual knowledge of the statute or specific intent to violate it; |
| • | the federal civil False Claims Act, which prohibits individuals or entities from, among other things, knowingly presenting, or causing to be presented, claims for payment of government funds that are false or fraudulent, and making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, a claim including items or services resulting from a violation of the AKS constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes civil and criminal liability for executing or attempting to execute a scheme to defraud any healthcare benefit program and creates federal criminal laws that prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; |
| • | the federal transparency requirements under the Health Care Reform Law, which require manufacturers of drugs, devices, biologics and medical supplies to report to the Department of Health and Human Services information related to payments and other transfers of value provided to physicians and teaching hospitals and physician ownership and investment interests; |
| • | federal and state data protection laws, including HIPAA, which imposes criminal penalties for improper acquisition of patient information. State data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., |
44
| Section 5 of the FTC Act), govern the collection, use, disclosure, and protection of health-related and other personal information. Failure to comply with data protection laws and regulations could result in government enforcement actions (which could include civil and/or criminal penalties), private litigation and/or adverse publicity and could negatively affect our operating results and business; and |
| • | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, that may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to implement compliance programs, comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or to track and report gifts, compensation and other remuneration provided to physicians and other health care providers; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal laws, thus complicating compliance efforts. |
Violations of any of these laws or any other governmental regulations that may apply to us, may subject us to significant civil, criminal and administrative sanctions including penalties, damages, fines, imprisonment, and exclusion from government funded healthcare programs, such as Medicare and Medicaid.
Employees
We currently have 193 full-time employees and 1 temporary employee. None of our employees are covered by collective bargaining agreements, and we consider relations with our employees to be good.
Facilities
Our principal executive offices are located at 490 Lapp Road, Malvern, PA 19355, where we occupy approximately 8,000 square feet of laboratory and office space. We have an office services agreement, or the Office Services Agreement, with Malvern Consulting Group, or MCG, a consulting company affiliated with our Chief Executive Officer, which includes the use of space as well as the use certain equipment and access to certain administrative services (for example, telephones, copy machines, kitchen facilities). We believe that this agreement is on arm’s length terms and is adequate for our current needs. The Office Services Agreement is on a month-to-month basis and we have provided notice of termination of this agreement effective December 31, 2016. In August 2016, we entered into a five-year lease, directly with the landlord of our Malvern facility with a lease term that will begin on January 1, 2017.
We currently own and operate a 97,000 square foot, DEA-licensed facility in Gainesville, Georgia.
Legal Proceedings
Other than as described in our most recent Annual Report on Form 10-K filed with the SEC, as revised or supplemented by our Quarterly Reports on Form 10-Q filed with the SEC since the filing of our most recent Annual Report on Form 10-K, each of which is incorporated by reference into this prospectus, we are not a party to any material litigation or proceeding and are not aware of any material litigation or proceeding, pending or threatened against us.
45
The following table sets forth our capitalization as of September 30, 2016:
| • | on an actual basis; and |
| • | on a pro forma basis to give effect to our sale of 500,000 shares of common stock for approximately $3.6 million in net proceeds under the Aspire Agreement between October 1, 2016 and November 22, 2016; and |
| • | on a pro forma as adjusted basis, giving effect to the pro forma adjustments set forth above, and to further reflect the sale by us of shares of our common stock in this offering at the public offering price of $ per share, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. |
The pro forma information below is illustrative only and our capitalization following the closing of this offering will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. You should read the data set forth in the table below in conjunction with our consolidated financial statements, including the related notes, and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” from our Quarterly Report on Form 10-Q for quarter ended September 30, 2016, which are incorporated by reference into this prospectus.
| (amounts in thousands, except share and per share data) | September 30, 2016 |
September 30, 2016 (pro forma) |
September 30, 2016 (pro forma as adjusted) |
|||||||||
| Cash and cash equivalents |
$ | 24,752 | 28,373 | $ | ||||||||
|
|
|
|
|
|
|
|||||||
| Debt (including current portion)(1) |
24,236 | 24,236 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Stockholders’ equity: |
||||||||||||
| Preferred stock, $0.01 par value. Authorized, 10,000,000 shares; none issued and outstanding, actual and as adjusted |
— | — | — | |||||||||
| Common stock, $0.01 par value, Authorized, 50,000,000 shares; 11,863,660 issued and outstanding, actual; 12,363,660 issued and outstanding, pro forma; and shares issued and outstanding, pro forma as adjusted |
119 | 124 | ||||||||||
| Additional paid-in capital |
91,378 | 94,994 | ||||||||||
| Accumulated deficit |
(50,866 | ) | (50,866 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Total shareholders’ equity |
40,631 | 44,252 | ||||||||||
|
|
|
|
|
|
|
|||||||
| Total capitalization |
$ | 64,867 | 68,488 | $ | ||||||||
|
|
|
|
|
|
|
|||||||
| (1) | Includes principal balance outstanding of $27,347, net of unamortized deferred issuance costs of $3,111. |
The foregoing table and calculations are based on 11,863,660 shares of our common stock outstanding as of September 30, 2016, and excludes:
| • | 2,343,819 shares of our common stock issuable upon the exercise of stock options outstanding as of September 30, 2016 at a weighted-average exercise price of $7.00 per share; |
| • | 19,975 shares of our common stock issuable upon the vesting and settlement of restricted stock units outstanding as of September 30, 2016; |
46
| • | 174 shares of our common stock available for future issuance as of September 30, 2016 under our 2008 Stock Option Plan; |
| • | 855,022 shares of our common stock available for future issuance as of September 30, 2016 under our Amended and Restated Equity Incentive Plan; |
| • | 784,928 shares of our common stock issuable upon the exercise of outstanding warrants as of September 30, 2016 with a weighted average exercise price of $12.05 per share; and |
| • | except with respect to the pro forma and pro forma as adjusted calculations, 500,000 shares of our common stock sold from October 1, 2016 to November 22, 2016 under the Aspire Agreement. |
47
If you invest in our common stock, your interest will be diluted immediately to the extent of the difference between the public offering price per share you will pay in this offering and the as adjusted net tangible book value per share of our common stock after this offering. Net tangible book value per share represents our total tangible assets less total liabilities, divided by the number of shares of our common stock outstanding.
As of September 30, 2016, our historical net tangible book value was $(4.2) million, or $(0.35) per share, based on 11,863,660 shares of common stock outstanding as of September 30, 2016. As of September 30, 2016, our pro forma net tangible book value was $(0.6) million, or $(0.05) per share, based on 11,863,660 shares of common stock outstanding as of September 30, 2016, and giving effect to our sale of 500,000 shares of common stock for approximately $3.6 million in net proceeds under the Aspire Agreement between October 1, 2016 and November 22, 2016. After giving effect to our issuance and sale of shares of common stock in this offering at the public offering price of $ per share, after deducting underwriting discounts and commissions and estimated offering expenses payable by us, the as adjusted net tangible book value as of September 30, 2016 would have been $ million, or $ per share. This represents an immediate increase in as adjusted net tangible book value to existing shareholders of $ per share and an immediate dilution to new investors purchasing common stock in this offering of $ per share.
The following table illustrates this per share dilution to the new investors purchasing shares of common stock in this offering:
| Public offering price per share |
$ | |||||||
| Historical net tangible book value per share at September 30, 2016 |
$ | (0.35 | ) | |||||
| Pro forma increase in net tangible book value per share attributable to sales of our common stock under the Aspire Agreement between October 1, 2016 and November 22, 2016 |
$ | 0.30 | ||||||
| Pro forma net tangible book value per share at September 30, 2016 |
$ | (0.05 | ) | |||||
| Increase in net tangible book value per share attributable to new investors purchasing shares in this offering |
||||||||
|
|
|
|
|
|||||
| Pro forma as adjusted net tangible book value per share after this offering |
||||||||
|
|
|
|||||||
| Dilution per share to new investors in this offering |
$ | |||||||
|
|
|
The foregoing table and calculations are based on 11,863,660 shares of our common stock outstanding as of September 30, 2016, and excludes:
| • | 2,343,819 shares of our common stock issuable upon the exercise of stock options outstanding as of September 30, 2016 at a weighted-average exercise price of $7.00 per share; |
| • | 19,975 shares of our common stock issuable upon the vesting and settlement of restricted stock units outstanding as of September 30, 2016; |
| • | 174 shares of our common stock available for future issuance as of September 30, 2016 under our 2008 Stock Option Plan; |
| • | 855,022 shares of our common stock available for future issuance as of September 30, 2016 under our Amended and Restated Equity Incentive Plan; |
| • | 784,928 shares of our common stock issuable upon the exercise of outstanding warrants as of September 30, 2016 with a weighted average exercise price of $12.05 per share; and |
48
| • | except with respect to the pro forma and pro forma as adjusted calculations, 500,000 shares of our common stock sold from October 1, 2016 to November 22, 2016 under the Aspire Agreement. |
To the extent that any options or warrants are exercised, new options are issued under our equity incentive plans or we otherwise issue additional shares of common stock in the future at a price less than the public offering price, there may be further dilution to new investors purchasing common stock in this offering.
49
CERTAIN RELATIONSHIPS AND TRANSACTIONS WITH RELATED PERSONS
Since January 1, 2013, we have engaged in the following transactions with our directors, executive officers, holders of more than 5% of our voting securities, and affiliates or immediate family members of our directors, executive officers, and holders of more than 5% of our voting securities. We believe that all of these transactions were on terms as favorable as could have been obtained from unrelated third parties.
Sales of Common Stock
On July 7, 2015, we closed a private placement with certain accredited investors, including Broadfin Capital, LLC, or Broadfin, which held more than 5% of our common stock at the time of the transaction. As part of the private placement, the Company sold 646,553 shares of common stock at a price per share of $11.60 to Broadfin and its affiliates for proceeds of $7.5 million. The Company sold an aggregate of 1,379,311 shares of common stock at a price per share of $11.60 in the private placement for net proceeds of $14.8 million.
On August 19, 2016, we closed an underwritten public offering in which we sold 1,986,666 shares of common stock at a price per share of $7.50 for net proceeds of approximately $13.4 million. Broadfin, which held more than 5% of our common stock at the time of the transaction, purchased 390,000 shares of common stock at a price of $7.50 per share.
Relationship with Malvern Consulting Group, Inc.
Ms. Henwood, our President and Chief Executive Officer, owns a majority of the stock of MCG, a pharmaceutical incubator and consulting firm. Thomas F. Henwood, Ms. Henwood’s husband, who is also a shareholder of our company, is a consultant for, and a shareholder of, MCG. In addition, Matthew Henwood, Ms. Henwood’s son, is the President of, and a shareholder of, MCG. Certain other employees of MCG are immediate family members of Ms. Henwood, including Christopher Sharr, Ms. Henwood’s brother, and Suzanne Sharr, Ms. Henwood’s sister-in-law. Since formation, we have entered into various transactions with MCG, as detailed below. Since becoming a public company we have sought to decrease our involvement with MCG and plan to continue to do so.
During 2013, 2014 and 2015, certain of our executive officers, Ms. Henwood, Mr. Mack, Ms. Myers, who is also Ms. Henwood’s sister, and Ms. Nichols were also employed by MCG. Following the termination from such employment of Ms. Nichols, Mr. Mack and Ms. Myers in March, September and October of 2015, respectively, Ms. Henwood, Mr. Mack and Ms. Myers provide minimal consulting services from time to time to MCG. We anticipate that no consulting services will be provided to MCG after December 31, 2016.
We are party to a Master Consulting Services Agreement with MCG. Pursuant to the agreement, MCG provides us with certain consulting services for a fee based upon hourly rates previously approved by our Audit Committee. In consideration for such services, we have recorded $0.3 million, $0.5 million and $0.5 million for the fiscal years ended December 31, 2013, 2014 and 2015, respectively, and we have recorded $0.3 million through September 30, 2016. A portion of these amounts are used to pay a portion of the respective salaries of MCG employees that, as described above, include immediate family members of Ms. Henwood. We expect to terminate this agreement effective December 31, 2016.
We are party to an Office Services Agreement with MCG for the lease of an aggregate of 8,458 square feet of office and lab space located at 490 Lapp Road, Malvern, Pa 19355 and the provision of IT services and general office support. Pursuant to the Office Services Agreement, we paid MCG $0.05 million, $0.1 million, and $0.1 million for the fiscal years ended December 31, 2013, 2014 and 2015, respectively, and we have paid $0.2 million through September 30, 2016. We expect to terminate
50
this agreement effective December 31, 2016. In August 2016, we entered into a five-year lease, directly with the landlord of our Malvern facility with a lease term that will begin on January 1, 2017.
Consulting Services of Ms. Henwood
From March 2013 until we entered into an employment agreement with Ms. Henwood in March 2014, we engaged Ms. Henwood to provide certain consulting services to our company. As part of such engagement, we paid Ms. Henwood a monthly fee of $12,500 in consideration for such services. Pursuant to such engagement, Ms. Henwood earned $0.1 million and $0.03 million during the fiscal years ended December 31, 2013 and 2014, respectively.
SCP Vitalife Partners II, L.P. and SCP Vitalife Partners (Israel) II, L.P.
SCP Vitalife Partners II, L.P. and SCP Vitalife Partners (Israel) II, L.P., as defined as SCP Vitalife, are venture capital funds that provided substantially all of our funding prior to our IPO. Ms. Henwood was a venture partner in SCP Vitalife Partners II, L.P. until April 2013; however, she maintains a financial interest in the fund by virtue of her prior investments. In addition, each of Mr. Churchill, Dr. Ludomirski and Mr. Weisman, members of our Board, are directors of the corporate general partner of the common general partner of SCP Vitalife Partners II, L.P. and SCP Vitalife Partners (Israel) II, L.P., and managing members of companies providing certain management services to them.
Since 2008, in exchange for contributions to the Company of $3.75 million, we issued to SCP Vitalife an aggregate amount of 1,875,000 shares of our Series A Redeemable Convertible Preferred Stock; and for contributions to the Company of $9.6 million, we issued to SCP Vitalife our 8% Convertible Promissory Notes. All of the shares of Series A Convertible Preferred Stock and 8% Convertible Promissory Notes issued to SCP Vitalife were converted to shares of our common stock upon consummation of our IPO.
We are also party to an Investor Rights Agreement with SCP Vitalife, which provides for, among other things, certain rights relating to the registration of their shares of common stock and the purchase of future securities sold by us. Except for the registration rights, all rights under this agreement terminated upon completion of our IPO. The registration rights will terminate on the third anniversary of our IPO, or earlier for any particular holder with registration rights when all securities held by that shareholder may be sold pursuant to Rule 144 under the Securities Act during any 90-day period.
Churchill Trust
The Churchill Trust, a trust for the benefit of Justin Churchill, the son of Mr. Churchill, a member of our Board, provided certain of our funding. From 2009 through our IPO in 2014, in exchange for aggregate contributions to us of $0.3 million, we issued an aggregate amount of 125,000 shares of our Series A Redeemable Convertible Preferred Stock to the trust which was converted to common stock at the time of the IPO.
51
The following table sets forth certain information regarding the beneficial ownership of Common Stock as of November 30, 2016 by (a) each person known by us to be the beneficial owner of more than 5% of the outstanding shares of Common Stock, (b) each named executive officer identified in the Summary Compensation Table below, (c) each director and nominee for director, and (d) all executive officers and directors as a group.
The percentage of Common Stock outstanding is based on 11,863,660 shares of our Common Stock outstanding as of November 30, 2016. For purposes of the table below, and in accordance with the rules of the SEC, we deem shares of Common Stock subject to options that are currently exercisable or exercisable within sixty days of November 30, 2016 to be outstanding and to be beneficially owned by the person holding the options for the purpose of computing the percentage ownership of that person, but we do not treat them as outstanding for the purpose of computing the percentage ownership of any other person. Except as otherwise noted, each of the persons or entities in this table has sole voting and investing power with respect to all of the shares of Common Stock beneficially owned by them, subject to community property laws, where applicable. Except as otherwise noted below, the street address of each beneficial owner is c/o Recro Pharma, Inc., 490 Lapp Road, Malvern, PA 19355.
| Shares Beneficially Owned Prior to Offering |
Shares Beneficially Owned After Offering |
|||||||||||||||
| Name of Beneficial Owner |
Number of Shares |
Percentage | Number of Shares |
Percentage | ||||||||||||
| 5% or Greater Shareholders | ||||||||||||||||
| Broadfin Capital, LLC(1) 300 Park Avenue New York, NY 10022 |
2,450,086 | 20.7 | ||||||||||||||
| SCP Vitalife Partners II, L.P.(2) 1200 Liberty Ridge Drive Suite 300 Wayne, PA 19087 |
2,322,824 | 19.6 | ||||||||||||||
| SCP Vitalife Partners (Israel) II, L.P.(2) 32B Habarzel St. Ramat Hachayal Tel Aviv 69710 Israel |
776,131 | 6.5 | ||||||||||||||
| Cormorant Asset Management, LLC(3) 100 High Street Boston, MA 02110 |
700,000 | 5.9 | ||||||||||||||
| Tourbillon Capital Partners, L.P.(4) 444 Madison Avenue, 26th Floor New York, NY 10022 |
646,552 | 5.4 | ||||||||||||||
| Named Executive Officers and Directors | ||||||||||||||||
| Gerri Henwood(5) |
452,913 | 3.8 | ||||||||||||||
| Randall Mack(6) |
117,227 | * | ||||||||||||||
| Stewart McCallum, M.D.(7) |
29,148 | * | ||||||||||||||
| Alfred Altomari(8) |
45,332 | * | ||||||||||||||
| William L. Ashton(9) |
57,332 | * | ||||||||||||||
| Michael Berelowitz(10) |
45,332 | * | ||||||||||||||
| Winston J. Churchill(11)(12) |
3,179,287 | 23.9 | ||||||||||||||
| Abraham Ludomirski(13) |
3,179,287 | 23.9 | ||||||||||||||
| Wayne Weisman(14) |
3,179,287 | 23.9 | ||||||||||||||
| Karen Flynn(15) |
16,666 | * | ||||||||||||||
| All executive officers and directors as a group (15 persons)(16) |
4,280,615 | 32.1 | ||||||||||||||
footnotes on following page
52
| * | Less than 1%. |
| (1) | Based upon information set forth in the Schedule 13D/A filed on August 11, 2016 and information set forth in Form 4 filed on August 17, 2016 by Broadfin, Broadfin Healthcare Master Fund, Ltd., or Master Fund, and Kevin Kotler. Broadfin, Master Fund and Mr. Kotler have shared voting and dispositive power over 2,450,086 shares of common stock. Broadfin and Mr. Kotler each disclaim beneficial ownership of the share reported herein except to the extent of its or his pecuniary interest therein. |
| (2) | Based upon information set forth in the Schedule 13D filed on March 21, 2014 and information set forth in Form 4’s filed on November 25, 2015, November 30, 2015, December 1, 2016, May 23, 2016 and May 25, 2016 by SCP Vitalife Partners II, L.P., or SCP Vitalife Partners, SCP Vitalife Partners (Israel) II, L.P., or SCP Vitalife Israel, SCP Vitalife II Associates, L.P., or SCP Vitalife Associates, SCP Vitalife II GP, LTD (SCP Vitalife GP), Winston J. Churchill, Jeffrey Dykan, Abraham Ludomirski, and Wayne B. Weisman. SCP Vitalife Partners beneficially owns 2,322,824 shares of common stock and SCP Vitalife Israel beneficially owns 776,131 shares of common stock. As the general partner of SCP Vitalife Partners and SCP Vitalife Israel, SCP Vitalife Associates may be deemed to beneficially own 3,098,955 shares of common stock. As the general partner of SCP Vitalife Associates, SCP Vitalife GP may be deemed to beneficially own 3,098,955 shares of common stock. As directors of SCP Vitalife GP, Messrs. Churchill, Dykan and Weisman and Dr. Ludomirski may be deemed to beneficially own 3,098,955 shares of common stock. SCP Vitalife Partners shares dispositive and voting power with respect to the 2,322,824 shares of common stock owned. SCP Vitalife Israel shares dispositive and voting power with respect to the 776,131 shares of common stock owned. SCP Vitalife Associates, SCP Vitalife GP, Messrs. Churchill, Dykan and Weisman and Dr. Ludomirski have shared dispositive and voting power with respect to the aggregate 3,098,955 shares of common stock owned by SCP Vitalife Partners and SCP Vitalife Israel. |
| (3) | Based upon information set forth in the Schedule 13G/A filed on February 17, 2015 by Cormorant Global Healthcare Master Fund, LP, or Cormorant, Cormorant Global Healthcare GP, LLC, or General Partner, Cormorant Asset Management, LLC, or Investment Manager and Bihua Chen. Cormorant Global Healthcare GP, LLC serves as the general partner and Cormorant Asset Management, LLC serves as the investment manager of Cormorant. Ms. Chen serves as the managing member of General Partner and Investment Manager. Cormorant, General Partner, Investment Manager and Ms. Chen share voting and dispositive power over all shares. Cormorant, General Partner, Investment Manager and Ms. Chen each disclaim beneficial ownership of the shares reported herein except to the extent of its or her pecuniary interest therein. |
| (4) | Based upon information set forth in the Schedule 13G filed on July 17, 2015 by Tourbillon Capital Partners, L.P., or Tourbillon, and Jason H. Karp. Mr. Karp serves as the Chief Executive Officer of Tourbillon. Tourbillon and Mr. Karp share voting and dispositive power over 646,552 shares of common stock. |
| (5) | Ms. Henwood holds 104,500 shares of our common stock and stock options to purchase 348,413 shares of our common stock that may be exercised within 60 days of November 30, 2016. Ms. Henwood’s husband, Thomas Henwood, holds 50,000 shares of our common stock. As spouses, Mr. and Ms. Henwood may be deemed to beneficially own the shares of our common stock that are held by the other spouse. Mr. and Ms. Henwood disclaim beneficial ownership of the shares of our common stock that are held by the other spouse. |
| (6) | Mr. Mack holds 5,554 shares of our common stock and stock options to purchase 111,673 shares of our common stock that may be exercised within 60 days of November 30, 2016. |
| (7) | Dr. McCallum holds 1,250 shares of our common stock and stock options to purchase 27,898 shares of our common stock that may be exercised within 60 days of November 30, 2016. These options were not granted pursuant to any of our equity compensation plans and were granted as an inducement grant pursuant to NASDAQ listing rule 5635(c)(4). |
| (8) | Mr. Altomari holds stock options to purchase 45,332 shares of our common stock that may be exercised within 60 days of November 30, 2016. |
| (9) | Mr. Ashton holds exercisable stock options to purchase 57,332 shares of our common stock that may be exercised within 60 days of November 30, 2016. |
| (10) | Dr. Berelowitz holds stock options to purchase 45,332 shares of our common stock that may be exercised within 60 days of November 30, 2016. |
| (11) | Mr. Churchill holds stock options to purchase 45,332 shares of our common stock that may be exercised within 60 days of November 30, 2016. Mr. Churchill has shared voting and investment power with respect to 3,098,955 shares of our common stock that are held by SCP Vitalife, of which he is a partner. |
| (12) | Mr. Churchill disclaims beneficial ownership of 50,000 shares of our common stock that are held by the Churchill Trust for the benefit of his son and stock options to purchase 33,200 shares of our common stock held by his son. |
| (13) | Dr. Ludomirski holds stock options to purchase 45,332 shares of our common stock that may be exercised within 60 days of November 30, 2016. Dr. Ludomirski has shared voting and investment power with respect to 3,098,955 shares of our common stock that are held by SCP Vitalife, of which he is a partner. |
| (14) | Mr. Weisman holds stock options to purchase 45,332 shares of our common stock that may be exercised within 60 days of November 30, 2016. Mr. Weisman has shared voting and investment power with respect to 3,098,955 shares of our common stock that are held by SCP Vitalife, of which he is a partner. |
| (15) | Ms. Flynn holds stock options to purchase 16,666 shares of our common stock that may be exercised within 60 days of November 30, 2016. |
| (16) | Includes stock options to purchase 970,956 shares of our common stock that may be exercised within 60 days of November 30, 2016. |
53
DESCRIPTION OF OUR CAPITAL STOCK
The following description of our capital stock and provisions of our articles of incorporation, bylaws and the Pennsylvania Business Corporation law are summaries and are qualified in their entirety by reference to the articles of incorporation and the bylaws.
Pursuant to our Second Amended and Restated Articles of Incorporation, our authorized capital stock consists of 50,000,000 shares of common stock, par value of $0.01 per share, and 10,000,000 shares of preferred stock, par value $0.01 per share, to be designated from time to time by our board.
Common Stock
As of November 30, 2016, there were 12,363,660 shares of our common stock issued and outstanding. Holders of our common stock are entitled to one vote for each share held on all matters submitted to a vote of shareholders, including the election of directors, and do not have cumulative voting rights. Accordingly, the holders of a majority of the outstanding shares of common stock in person or represented by proxies in any election of directors can elect all of the directors standing for election, if they so choose, other than any directors that holders of any preferred stock that we may issue may be entitled to elect.
Subject to preferences that may be applicable to any then-outstanding shares of preferred stock, holders of our common stock are entitled to receive ratably dividends when, as, and if declared by our board of directors out of funds legally available therefor, subject to any preferential dividend rights of outstanding preferred stock. In the event of our liquidation, dissolution, or winding up, holders of our common stock will be entitled to ratably receive the net assets of our company available after the payments of all debts and other liabilities and subject to the prior rights of the holders of any then-outstanding shares of preferred stock.
Holders of our common stock have no preemptive, subscription, redemption or conversion rights. All outstanding shares of our common stock are, and the common stock to be outstanding upon completion of this offering, will be, duly authorized, validly issued, fully paid and non- assessable. The rights and privileges of the holders of the common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock that we may designate and issue in the future.
Preferred Stock
Our board of directors has the authority, without further action by our shareholders, to issue up to 10,000,000 shares of preferred stock in one or more series, to establish from time to time the number of shares to be included in each such series, to fix the dividend, voting and other rights, preferences and privileges of the shares of each wholly unissued series and any qualifications, limitations or restrictions thereon, and to increase or decrease the number of shares of any such series, but not below the number of shares of such series then outstanding. Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of our common stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in our control and may adversely affect the market price of the common stock and the voting and other rights of the holders of our common stock. We have no current plans to issue any shares of preferred stock.
Common Stock Warrants
We issued to the representatives of the underwriters in our IPO warrants to purchase up to 150,000 shares of our common stock, with a per share exercise price equal to $12.00, or 150% of the public
54
offering price, or IPO warrants. The IPO warrants provide for certain piggyback registration rights. The IPO warrants are exercisable by the underwriters at any time, in whole or in part, until March 12, 2019. On July 22, 2015, we issued 2,941 shares of our common stock pursuant to a cashless exercise of 10,000 IPO warrants with an exercise price of $12.00 per share.
In connection with the Gainesville Transaction, we issued to Alkermes a seven-year warrant to purchase an aggregate of 350,000 shares of our common stock, with an exercise price of $19.46 per share. In addition, we issued to OrbiMed a seven year warrant to purchase an aggregate of 294,928 shares of our common stock, with an exercise price of $3.28 per share, subject to certain adjustments.
Registration Rights
Private Placement
In July 2015 we completed a private placement to certain investors of 1,379,311 shares of our common stock. In connection with the private placement, we entered into a securities purchase agreement, or Purchase Agreement, with such investors under which we granted the investors certain registration rights with respect to the shares purchased. In particular, the Purchase Agreement required us to file a registration statement with the SEC to register the sale of such shares within 45 days of the consummation of the private placement and to maintain continuous effectiveness of the registration statement. A registration statement relating to such shares was filed on August 20, 2015 and declared effective by the SEC on September 1, 2015.
IPO Warrants
As stated above, holders of the IPO warrants have certain piggyback registration rights. If at any time prior to the third anniversary of our IPO we propose to register any shares of our common stock under the Securities Act, subject to certain exceptions, the holders of the IPO warrants will be entitled to notice of the registration and the right to include the shares of common stock issuable upon exercise of their IPO warrants in the registration statement. If our proposed registration involves an underwriting, the underwriter of such offering will have the right to limit the number of shares to be underwritten for reasons related to the marketing of the shares.
Prior Holders of Series A Redeemable Convertible Preferred Stock
Holders of our common stock that were issued upon conversion of our Series A Redeemable Convertible Preferred Stock immediately prior to the closing of our IPO are entitled to the following rights with respect to the registration of such shares, or registrable securities, for public resale under the Securities Act, pursuant to an Investor Rights Agreement by and among us and certain of our shareholders. The registration of shares of common stock as a result of the following rights being exercised would enable holders to trade these shares without restriction under the Securities Act when the applicable registration statement is declared effective.
Demand Registration Rights. If at any time the holders of a majority of the registrable securities request in writing that we file a registration statement under the Securities Act for the registration of at least 20% of our common stock then outstanding with an aggregate price of at least $20 million, we may be required to register their shares. We are obligated to effect no more than two registrations for the holders of registrable securities in response to these demand registration rights. If the holders requesting registration intend to distribute their shares by means of an underwriting, the underwriter of such offering will have the right to limit the numbers of shares to be underwritten on a pro rata basis for reasons related to the marketing of the shares.
Piggyback Registration Rights. If at any time we propose to register any shares of our common stock under the Securities Act, subject to certain exceptions, the holders of registrable securities will be entitled
55
to notice of the registration and the right to include their shares of registrable securities in the registration statement. If our proposed registration involves an underwriting, the underwriter of such offering will have the right to limit the number of shares to be underwritten for reasons related to the marketing of the shares.
Form S-3 Registration Rights. If at any time we are entitled under the Securities Act to register our shares of common stock on Form S-3, holders of not less than 10% of the registrable securities then outstanding request in writing that we register their shares for public resale on Form S-3 and the reasonably anticipated price to the public is $10 million or more, we will be required to use commercially reasonable efforts to effect such registration; provided, however, that we will not be required to effect such a registration if (1) we certify in a certificate signed by our Chief Executive Officer that we intend to engage in a registered public offering within 90 days of receiving the Form S-3 request, or (2) we certify in a certificate signed by our Chief Executive Officer stating that in our good faith judgment, it would be detrimental to us for such registration on Form S-3 to be effected at such time, in which event we have the right to defer the filing of the Form S-3 registration statement for a period of not more than 120 days.
Expenses. Subject to certain exceptions, and other than underwriting discounts and selling commissions, we will be required to pay all expenses incurred by us related to any registration effected pursuant to the exercise of these registration rights. These expenses may include all registration and filing fees, printing expenses, fees and disbursements of our counsel, blue sky fees and expenses and the expenses of any special audits incident to or required by the registration.
Termination of Registration Rights. These registration rights terminate on March 12, 2017. In addition, a holder’s registration rights will expire if all registrable securities held by and issuable to such holder could be sold under Rule 144 of the Securities Act during any 90-day period.
All applicable registration rights with respect to the registration statement of which this prospectus forms a part have been waived.
Anti-Takeover Effects of Pennsylvania Law and Our Articles of Incorporation and Bylaws
Pennsylvania Anti-Takeover Law
Provisions of the Pennsylvania Business Corporation Law of 1988, or the PBCL, applicable to us provide, among other things, that:
| • | we may not engage in a business combination with an “interested shareholder,” generally defined as a holder of 20% of a corporation’s voting stock, during the five-year period after the interested shareholder became such except under certain specified circumstances; |
| • | holders of our common stock may object to a “control transaction” involving us (a control transaction is defined as the acquisition by a person or group of persons acting in concert of at least 20% of the outstanding voting stock of a corporation), and demand that they be paid a cash payment for the “fair value” of their shares from the “controlling person or group”; |
| • | holders of “control shares” will not be entitled to voting rights with respect to any shares in excess of specified thresholds, including 20% voting control, until the voting rights associated with such shares are restored by the affirmative vote of a majority of disinterested shares and the outstanding voting shares of the Company; and |
| • | any “profit,” as defined, realized by any person or group who is or was a “controlling person or group” with respect to us from the disposition of any equity securities of within 18 months after the person or group became a “controlling person or group” shall belong to and be recoverable by us. |
56
Pennsylvania-chartered corporations may exempt themselves from these and other anti-takeover provisions. Our articles of incorporation do not provide for exemption from the applicability of these or other anti-takeover provisions in the PBCL.
The provisions noted above may have the effect of discouraging a future takeover attempt that is not approved by our board of directors but which individual shareholders may consider to be in their best interests or in which shareholders may receive a substantial premium for their shares over the then current market price. As a result, shareholders who might wish to participate in such a transaction may not have an opportunity to do so. The provisions may also result in the removal of our board of directors or management more difficult. Furthermore, such provisions could render our company being deemed less attractive to a potential acquiror and/or could result in our shareholders receiving a lesser amount of consideration for their shares of our common stock than otherwise could have been available either in the market generally and/or in a takeover.
Articles of Incorporation and Bylaws
Provisions of our articles of incorporation and bylaws may delay or discourage transactions involving an actual or potential change of control or change in our management, including transactions in which shareholders might otherwise receive a premium for their shares, or transactions that our shareholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our articles of incorporation and bylaws:
| • | divide our board of directors into three classes with staggered three-year terms; |
| • | provide that a special meeting of shareholders may be called only by a majority of our board of directors; |
| • | establish advance notice procedures with respect to shareholder proposals to be brought before a shareholder meeting and the nomination of candidates for election as directors, other than nominations made by or at the direction of the board of directors or a committee of the board of director; |
| • | provide that shareholders may only act at a duly organized meeting; and |
| • | provide that members of our board of directors may be removed from office by our shareholders only for cause by the affirmative vote of 75% of the total voting power of all shares entitled to vote generally in the election of directors. |
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Broadridge Corporate Issuer Solutions, Inc.
Stock Market Listing
Our shares of common stock are listed for trading on the NASDAQ Capital Market under the symbol “REPH.”
57
Piper Jaffray & Co., or Piper Jaffray, is acting as representative of each of the underwriters named below. Subject to the terms and conditions set forth in an underwriting agreement among us and the underwriters, we have agreed to sell to the underwriters, and each of the underwriters has agreed, severally and not jointly, to purchase from us, the number of shares of our common stock set forth opposite its name below.
| UNDERWRITER |
NUMBER OF SHARES |
|||
| Piper Jaffray & Co. |
||||
| Total |
||||
|
|
|
|||
Subject to the terms and conditions set forth in the underwriting agreement, the underwriters have agreed, severally and not jointly, to purchase all of the shares sold under the underwriting agreement if any of these shares are purchased. If an underwriter defaults, the underwriting agreement provides that the purchase commitments of the non-defaulting underwriters may be increased or the underwriting agreement may be terminated.
We have agreed to indemnify the several underwriters against certain liabilities, including liabilities under the Securities Act relating to losses or claims resulting from material misstatements in or omissions from this prospectus supplement, the registration statement of which this prospectus is a part, certain free writing prospectuses that may be used in the offering and in any marketing materials used in connection with this offering and to contribute to payments the underwriters may be required to make in respect of those liabilities.
The underwriters are offering the shares, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel, including the validity of the shares, and other conditions contained in the underwriting agreement, such as the receipt by the underwriters of officers’ certificates and legal opinions. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
Commissions and Discounts
The representative has advised us that the underwriters propose initially to offer the shares to the public at the public offering price set forth on the cover page of this prospectus and to dealers at that price less a concession not in excess of $ per share. After the initial offering, the public offering price, concession or any other term of this offering may be changed.
We have granted to the underwriters an option, exercisable for 30 days from the date of this prospectus, to purchase up to additional shares of our common stock at the public offering price listed on the cover page of this prospectus, less underwriting discounts and commissions. To the extent the option is exercised, each underwriter will become obligated, subject to certain conditions, to purchase approximately the same percentage of the additional shares of common stock as the number listed next to the underwriter’s name in the preceding table bears to the total number of shares of common stock listed next to the names of all underwriters in the table above.
58
The following table shows the public offering price, underwriting discounts and commissions and proceeds before expenses to us. The information assumes either no exercise or full exercise by the underwriters of their option to purchase additional shares.
| Per Share | Without Option |
With Option |
||||||||||
| Public offering price |
$ | $ | $ | |||||||||
| Underwriting discounts and commissions paid by us |
$ | $ | $ | |||||||||
| Proceeds to us, before expenses |
$ | $ | $ | |||||||||
The estimated offering expenses payable by us, exclusive of the underwriting discounts and commissions, are approximately $ . We have also agreed to reimburse the underwriters for certain of their expenses in an amount not to exceed $160,000, including expenses of up to $25,000, in connection with the clearance of this offering with the Financial Industry Regulatory Authority, as set forth in the underwriting agreement.
Our common stock is listed on The Nasdaq Capital Market under the trading symbol “REPH.”
No Sales of Similar Securities
We and each of our directors and executive officers and some of our stockholders have agreed that we and they will not, without the prior written consent of Piper Jaffray, subject to certain limited exceptions, directly or indirectly:
| • | offer, pledge, announce the intention to sell, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, make any short sale or otherwise transfer or dispose of, directly or indirectly, any shares of common stock or any securities convertible into, exercisable or exchangeable for or that represent the right to receive common stock (including without limitation, common stock which may be deemed to be beneficially owned by the holder in accordance with the rules and regulations of the SEC and securities which may be issued upon exercise of a stock option or warrant) whether now owned or hereafter acquired; |
| • | enter into any swap or other agreement that transfers, in whole or in part, any of the economic consequences of ownership of the holder’s securities; |
| • | make any demand for or exercise any right with respect to, the registration of any common stock or any security convertible into or exercisable or exchangeable for common stock in each case that would require us to file a registration statement with the next 90 days of the date of the lock-up agreement; or |
| • | publicly disclose the intention to do any of the foregoing, |
for a period of 90 days after the public offering date set forth in this prospectus. However, in the case of our directors, executive officers and stockholders subject to the 90-day restricted period, these restrictions will not apply to transfers of our common stock or any security convertible into or exercisable for our common stock: (i) as a bona fide gift or gifts made by the holder, (ii) to any trust for the direct or indirect benefit of the holder or the holder’s immediate family, (iii) if the holder is a corporation, partnership, limited liability company, trust or other business entity, (1) to another corporation, partnership, limited liability company, trust or other business entity that is a direct or indirect affiliate (as defined in Rule 405 promulgated under the Securities Act) of the holder or (2) as a distribution to the holder’s limited partners, limited liability company members or stockholders, (iv) if the holder is a trust, to the beneficiary of such trust, (v) upon death by will or intestate succession, or (vi) pursuant to a qualified domestic relations order; provided, that (x) such transfers do not involve a disposition for value, (y) the transferee agrees in writing to be bound to the 90-day restricted period for
59
subsequent transfers, and (z) no filing by any party under Section 16(a) of the Exchange Act is required or shall be made voluntarily in connection with such transfer during the 90-day restricted period. In addition the restrictions will also not apply to (i) the exercise, conversion or exchange of any options or other convertible securities outstanding on the date the lockup agreement was signed (including by “net” or “cashless” exercise effected by the delivery or sale of the holder’s securities to us to the extent permitted by the instruments representing such options or other convertible securities and including the transfer of shares of common stock to us to satisfy tax withholding obligations in connection therewith), provided that no filing under Section 16(a) of the Exchange Act by any party is required or will be voluntarily made in connection with such exercise, conversion or exchange that reports a disposition of shares of common stock, except to report any transfer of shares of common stock to us to finance a “cashless” exercise or to satisfy tax withholding obligations as described above, and provided further, that such restrictions shall apply to any of the holder’s securities issued upon such exercise, conversion or exchange, or (ii) the establishment of any contract, instruction or plan that satisfies all of the requirements of Rule 10b5-1(c)(1)(i)(B) under the Exchange Act; provided, that no sales of the holder’s securities shall be made pursuant to such a plan prior to the expiration of the 90-day restricted period, and such a plan may only be established if no public announcement of the establishment or existence thereof, and no filing with the SEC or other regulatory authority in respect thereof or transactions thereunder or contemplated thereby, is required or made voluntarily by the holder, us or any other person during the 90-day restricted period. The restrictions also do not apply to any securities acquired by a holder in the open market after the date of the lock-up agreement, provided that no filing under Section 16(a) of the Exchange Act is required or will be voluntarily made in connection with any subsequent sale, transfer, gift or disposition.
During the 90-day restricted period, we may issue securities (a) to our directors, officers, employees and consultants pursuant to our employee benefit plans, equity incentive plans and other employee compensation plans existing on the date of the underwriting agreement and described in this prospectus and the documents incorporated herein; (b) pursuant to the exercise, exchange or conversion of any options, warrants, restricted stock units, rights or convertible securities outstanding on the date of the underwriting agreement, (c) in connection with any joint venture, commercial or collaborative relationship, the acquisition or license by us of the securities, businesses, property or other assets of another person, or an equity investment in the common stock by a non-financial investor as part of any such transaction, so long as each recipient of any such shares or other securities agrees to be bound by the terms of the 90-day restricted period, (d) pursuant to the Common Stock Purchase Agreement, dated as of February 2, 2015, between the Company and Aspire Capital Fund, LLC, in connection with an “at-the-market” equity offering, so long as such issuances and sales occur no earlier than 45 days after the date of the underwriting agreement and do not exceed an aggregate of $2.2 million in gross proceeds, and (f) to one or more non-financial investors in connection with an equity investment in the Company, as long as such issuances and sales occur no earlier than 45 days after the date of the underwriting agreement.
Piper Jaffray may, in its sole discretion and at any time or from time to time before the termination of the 90-day restricted period, release all or any portion of the securities subject to lock-up agreements. There are no existing agreements between the underwriters and any of our stockholders who will execute a lock-up agreement providing consent to the sale of shares prior to the expiration of the 90-day restricted period.
Price Stabilization, Short Positions and Penalty Bids
Until the distribution of the shares is completed, SEC rules may limit underwriters and selling group members from bidding for and purchasing shares of our common stock. However, the representatives may engage in transactions that stabilize the price of our common stock, such as bids or purchases to peg, fix or maintain that price.
60
In connection with this offering, the underwriters may purchase and sell shares of our common stock in the open market. These transactions may include short sales, purchases on the open market to cover positions created by short sales and stabilizing transactions. Short sales involve the sale by the underwriters of a greater number of shares than they are required to purchase in this offering. “Covered” short sales are sales made in an amount not greater than the underwriters’ option to purchase additional shares described above. The underwriters may close out any covered short position by either exercising their option to purchase additional shares or purchasing shares in the open market. In determining the source of shares to close out the covered short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the option to purchase additional shares. “Naked” short sales are sales in excess of the option to purchase additional shares. The underwriters must close out any naked short position by purchasing shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there may be downward pressure on the price of our common stock in the open market after pricing that could adversely affect investors who purchase in this offering. Stabilizing transactions consist of various bids for or purchases of shares of our common stock made by the underwriters in the open market prior to the closing of this offering.
The underwriters may also impose a penalty bid. This occurs when a particular underwriter repays to the underwriters a portion of the underwriting discount received by it because the representatives have repurchased shares sold by or for the account of such underwriter in stabilizing or short covering transactions.
Similar to other purchase transactions, the underwriters’ purchases to cover the syndicate short sales may have the effect of raising or maintaining the market price of our common stock or preventing or retarding a decline in the market price of our common stock. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market. The underwriters may conduct these transactions on NASDAQ, in the over-the-counter market or otherwise.
Neither we nor any of the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of our common stock. In addition, neither we nor any of the underwriters make any representation that the representatives will engage in these transactions or that these transactions, once commenced, will not be discontinued without notice.
Electronic Offer, Sale and Distributions of Shares
In connection with this offering, certain of the underwriters or securities dealers may distribute prospectuses by electronic means, such as e-mail. In addition, one or more of the underwriters may facilitate Internet distribution for this offering to certain of their Internet subscription customers. Any such underwriter may allocate a limited number of shares for sale to its online brokerage customers. An electronic prospectus is available on the Internet websites maintained by any such underwriter. Other than the prospectus in electronic format, the information on the websites of any such underwriter is not part of this prospectus.
Other Relationships
The underwriters and their respective affiliates are full service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities. Certain of the underwriters and their affiliates have engaged in, and may in the future engage in, investment banking and other commercial dealings in the ordinary course of business with us or our affiliates. They have received, or may in the future receive, customary fees and commissions for these transactions.
61
In the ordinary course of their various business activities, the underwriters and their respective affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the accounts of their customers, and such investment and securities activities may involve securities and/or instruments of the issuer. The underwriters and their respective affiliates may also make investment recommendations and/or publish or express independent research views in respect of such securities or instruments and may at any time hold, or recommend to clients that they acquire, long and/or short positions in such securities and instruments.
Selling Restrictions
European Economic Area
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (each, a Relevant Member State) an offer to the public of any shares of our common stock may not be made in that Relevant Member State, except that an offer to the public in that Relevant Member State of any shares of our common stock may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
| (a) | to any legal entity which is a qualified investor as defined in the Prospectus Directive; |
| (b) | to fewer than 100 or, if the Relevant Member State has implemented the relevant provision of the 2010 PD Amending Directive, 150, natural or legal persons (other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to obtaining the prior consent of the representatives for any such offer; or |
| (c) | in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of shares of our common stock shall result in a requirement for the publication by us or any underwriter of a prospectus pursuant to Article 3 of the Prospectus Directive. |
For the purposes of this provision, the expression an “offer to the public” in relation to any shares of our common stock in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any shares of our common stock to be offered so as to enable an investor to decide to purchase any shares of our common stock, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State, the expression “Prospectus Directive” means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending Directive, to the extent implemented in the Relevant Member State), and includes any relevant implementing measure in the Relevant Member State, and the expression “2010 PD Amending Directive” means Directive 2010/73/EU.
United Kingdom
Each underwriter has represented and agreed that:
| (a) | it has only communicated or caused to be communicated and will only communicate or cause to be communicated an invitation or inducement to engage in investment activity (within the meaning of Section 21 of the Financial Services and Markets Act 2000 (the FSMA)) received by it in connection with the issue or sale of the shares of our common stock in circumstances in which Section 21(1) of the FSMA does not apply to us; and |
| (b) | it has complied and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the shares of our common stock in, from or otherwise involving the United Kingdom. |
62
Canada
The shares of our common stock may be sold only to purchasers purchasing, or deemed to be purchasing, as principal that are accredited investors, as defined in National Instrument 45-106 Prospectus Exemptions or subsection 73.3(1) of the Securities Act (Ontario), and are permitted clients, as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale of the shares of our common stock must be made in accordance with an exemption from, or in a transaction not subject to, the prospectus requirements of applicable securities laws.
Securities legislation in certain provinces or territories of Canada may provide a purchaser with remedies for rescission or damages if the prospectus (including any amendment thereto) contains a misrepresentation, provided that the remedies for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser’s province or territory. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser’s province or territory for particulars of these rights or consult with a legal advisor.
Pursuant to section 3A.3 (or, in the case of securities issued or guaranteed by the government of a non-Canadian jurisdiction, section 3A.4) of National Instrument 33-105 Underwriting Conflicts (NI 33-105), the underwriters are not required to comply with the disclosure requirements of NI 33-105 regarding underwriter conflicts of interest in connection with this offering.
Hong Kong
The common shares may not be offered or sold in Hong Kong by means of any document other than (i) in circumstances which do not constitute an offer to the public within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong), or (ii) to “professional investors” within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder, or (iii) in other circumstances which do not result in the document being a “prospectus” within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong) and no advertisement, invitation or document relating to the shares may be issued or may be in the possession of any person for the purpose of issue (in each case whether in Hong Kong or elsewhere), which is directed at, or the contents of which are likely to be accessed or read by, the public in Hong Kong (except if permitted to do so under the laws of Hong Kong) other than with respect to common shares which are or are intended to be disposed of only to persons outside Hong Kong or only to “professional investors” within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder.
Singapore
This prospectus has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, this prospectus and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of the common shares may not be circulated or distributed, nor may the common shares be offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (i) to an institutional investor under Section 274 of the Securities and Futures Act, Chapter 289 of Singapore (the SFA), (ii) to a relevant person pursuant to Section 275(1), or any person pursuant to Section 275(1A), and in accordance with the conditions specified in Section 275 of the SFA or (iii) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the SFA, in each case subject to compliance with conditions set forth in the SFA.
Where the common shares are subscribed or purchased under Section 275 of the SFA by a relevant person which is:
| (a) | a corporation (which is not an accredited investor (as defined in Section 4A of the SFA)) the sole business of which is to hold investments and the entire share capital of which is owned by one or more individuals, each of whom is an accredited investor; or |
63
| (b) | a trust (where the trustee is not an accredited investor) whose sole purpose is to hold investments and each beneficiary of the trust is an individual who is an accredited investor, |
shares, debentures and units of shares and debentures of that corporation or the beneficiaries’ rights and interest (howsoever described) in that trust shall not be transferred within six months after that corporation or that trust has acquired the common shares pursuant to an offer made under Section 275 of the SFA except:
| (a) | to an institutional investor (for corporations, under Section 274 of the SFA) or to a relevant person defined in Section 275(2) of the SFA, or to any person pursuant to an offer that is made on terms that such shares, debentures and units of shares and debentures of that corporation or such rights and interest in that trust are acquired at a consideration of not less than $200,000 (or its equivalent in a foreign currency) for each transaction, whether such amount is to be paid for in cash or by exchange of securities or other assets, and further for corporations, in accordance with the conditions specified in Section 275 of the SFA; |
| (b) | where no consideration is or will be given for the transfer; or |
| (c) | where the transfer is by operation of law. |
Switzerland
The common shares may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (the SIX) or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering or marketing material relating to the common shares or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to the offering, or the common shares have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of common shares will not be supervised by, the Swiss Financial Market Supervisory Authority FINMA, and the offer of common shares has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes (CISA). Accordingly, no public distribution, offering or advertising, as defined in CISA, its implementing ordinances and notices, and no distribution to any non-qualified investor, as defined in CISA, its implementing ordinances and notices, shall be undertaken in or from Switzerland, and the investor protection afforded to acquirers of interests in collective investment schemes under CISA does not extend to acquirers of common shares.
United Arab Emirates
This offering has not been approved or licensed by the Central Bank of the United Arab Emirates (the UAE), Securities and Commodities Authority of the UAE and/or any other relevant licensing authority in the UAE including any licensing authority incorporated under the laws and regulations of any of the free zones established and operating in the territory of the UAE, in particular the Dubai Financial Services Authority (DFSA), a regulatory authority of the Dubai International Financial Centre (DIFC). The offering does not constitute a public offer of securities in the UAE, DIFC and/or any other free zone in accordance with the Commercial Companies Law, Federal Law No 8 of 1984 (as amended), DFSA Offered Securities Rules and NASDAQ Dubai Listing Rules, accordingly, or otherwise. The common shares may not be offered to the public in the UAE and/or any of the free zones.
64
The common shares may be offered and issued only to a limited number of investors in the UAE or any of its free zones who qualify as sophisticated investors under the relevant laws and regulations of the UAE or the free zone concerned.
France
This prospectus (including any amendment, supplement or replacement thereto) is not being distributed in the context of a public offering in France within the meaning of Article L. 411-1 of the French Monetary and Financial Code (Code monétaire et financier).
This prospectus has not been and will not be submitted to the French Autorité des marchés financiers (the AMF) for approval in France and accordingly may not and will not be distributed to the public in France.
Pursuant to Article 211-3 of the AMF General Regulation, French residents are hereby informed that:
| (a) | the transaction does not require a prospectus to be submitted for approval to the AMF; |
| (b) | persons or entities referred to in Point 2°, Section II of Article L.411-2 of the Monetary and Financial Code may take part in the transaction solely for their own account, as provided in Articles D. 411-1, D. 734-1, D. 744-1, D. 754-1 and D. 764-1 of the Monetary and Financial Code; and |
| (c) | the financial instruments thus acquired cannot be distributed directly or indirectly to the public otherwise than in accordance with Articles L. 411-1, L. 411-2, L. 412-1 and L. 621-8 to L. 621-8-3 of the Monetary and Financial Code. |
This prospectus is not to be further distributed or reproduced (in whole or in part) in France by the recipients of this prospectus. This prospectus has been distributed on the understanding that such recipients will only participate in the issue or sale of our common stock for their own account and undertake not to transfer, directly or indirectly, our common stock to the public in France, other than in compliance with all applicable laws and regulations and in particular with Articles L. 411-1 and L. 411-2 of the French Monetary and Financial Code.
65
The validity of the shares of common stock being offered by this prospectus will be passed upon for us by Hogan Lovells US LLP. Dechert LLP is counsel for the underwriters in connection with this offering.
The consolidated financial statements of Recro Pharma, Inc. as of December 31, 2015 and 2014, and for the years then ended, have been incorporated by reference herein in reliance upon the report of KPMG LLP, independent registered public accounting firm, incorporated by reference herein, and upon the authority of said firm as experts in accounting and auditing.
The audited combined historical financial statements of DARA included on page 6 of Recro Pharma, Inc.’s Current Report on Form 8-K/A dated June 2, 2015 have been so incorporated in reliance on the report of PricewaterhouseCoopers LLP, independent accountants, given on the authority of said firm as experts in auditing and accounting.
66
The SEC allows us to “incorporate by reference” information into this prospectus, which means that we can disclose important information to you by referring you to another document filed separately with the SEC. The SEC file number for the documents incorporated by reference in this prospectus is 001-36329. The documents incorporated by reference into this prospectus contain important information that you should read about us.
The following documents are incorporated by reference into this document:
| • | our Annual Report on Form 10-K for the year ended December 31, 2015, filed on March 24, 2016; |
| • | our Quarterly Reports on Form 10-Q for the periods ended March 31, 2016, June 30, 2016 and September 30, 2016, filed on May 12, 2016, August 11, 2016 and November 10, 2016, respectively; |
| • | those portions of our Definitive Proxy Statement on Schedule 14A filed on April 12, 2016 that are deemed “filed” with the SEC; and |
| • | our Current Reports on Form 8-K (other than portions thereof furnished under Item 2.02 or Item 7.01 of Form 8-K and exhibits accompanying such reports that relate to such items) filed with the SEC on April 16, 2015 (as amended by Form 8-K/A filed on June 2, 2015, Form 8-K/A filed on June 26, 2015, Form 8-K/A filed on July 21, 2015, Form 8-K/A filed on December 23, 2015 and Form 8-K/A filed on April 29, 2016), January 6, 2016, January 11, 2016, January 27, 2016, February 1, 2016, February 12, 2016, February 16, 2016, March 14, 2016, May 25, 2016, July 5, 2016, July 14, 2016, July 27, 2016, August 12, 2016, August 17, 2016, September 8, 2016, September 12, 2016, November 15, 2016, November 28, 2016 and November 30, 2016. |
We also incorporate by reference into this prospectus all documents (other than current reports furnished under Item 2.02 or Item 7.01 of Form 8-K and exhibits filed on such form that are related to such items) that are filed by us with the SEC pursuant to Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act after the date of the initial registration statement of which this prospectus is a part and prior to the effectiveness of such registration statement and all documents that are filed by us with the SEC pursuant to Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act after the date of this prospectus but prior to the termination of the offering. These documents include periodic reports, such as Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K, as well as proxy statements.
Any statement contained herein or in a document incorporated or deemed to be incorporated by reference into this document will be deemed to be modified or superseded for purposes of the document to the extent that a statement contained in this document or any other subsequently filed document that is deemed to be incorporated by reference into this document modifies or supersedes the statement.
You may request, orally or in writing, a copy of any or all of the documents incorporated herein by reference. These documents will be provided to you at no cost, by contacting: Susan Kim, Argot Partners, 767 Third Avenue, 29th Floor, New York, NY 10017, (212) 600-1902, email address:susan@argotpartners.com. In addition, copies of any or all of the documents incorporated herein by reference may be accessed at our website at http://www.recropharma.com. The information on such website is not incorporated by reference and is not a part of this prospectus.
67
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act, with respect to the shares of common stock being offered by this prospectus. This prospectus does not contain all of the information in the registration statement and its exhibits. For further information with respect to us and the common stock offered by this prospectus, we refer you to the registration statement and its exhibits. Statements contained in this prospectus as to the contents of any contract or any other document referred to are not necessarily complete, and in each instance, we refer you to the copy of the contract or other document filed as an exhibit to the registration statement. Each of these statements is qualified in all respects by this reference. You should rely only on information contained in, or incorporated by reference into, this prospectus. We have not authorized anyone to provide you with information different from that contained in this prospectus or incorporated by reference in this prospectus.
We file annual, quarterly and current reports, proxy statements and other information with the SEC. You may read and copy any document we file with the SEC at the SEC’s public reference room at 100 F Street NE, Room 1580, Washington, D.C. 20549. You may obtain information on the operation of the SEC’s public reference facilities by calling the SEC at 1-800-SEC-0330. You can request copies of these documents, upon payment of a duplicating fee, by writing to the SEC at its principal office at 100 F Street NE, Room 1580, Washington, D.C. 20549-1004. The SEC maintains an Internet website at http://www.sec.gov that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. Our SEC filings are accessible through the Internet at that website. Our reports on Forms 10-K, 10-Q and 8-K, and amendments to those reports, are also available for download, free of charge, as soon as reasonably practicable after these reports are filed with the SEC, at our website at www.recropharma.com. The content contained in, or that can be accessed through, our website is not a part of this prospectus.
68
Shares
Common Stock
PROSPECTUS
, 2016
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table sets forth all expenses, other than underwriting discounts and commissions, paid or payable by us in connection with this offering. All amounts shown are estimates except for the SEC registration fee and the Financial Industry Regulatory Authority, or FINRA, filing fee.
| Amount to be paid |
||||
| SEC registration fee |
$ | 6,664.25 | ||
| FINRA filing fee |
* | |||
| Printing expenses |
* | |||
| Accounting fees and expenses |
* | |||
| Legal fees and expenses |
* | |||
| Miscellaneous |
* | |||
|
|
|
|||
| Total |
$ | * | ||
| * | To be filed by amendment. |
Item 14. Indemnification of Directors and Officers.
The Company’s by-laws provide that, to the fullest extent permitted by Pennsylvania law, any officer or director of the Company who was or is a party or is threatened to be made a party to, any threatened, or pending or completed action, suit or proceeding, whether civil, criminal, administrative, or investigative, by reason of fact that he/she is or was acting as was a representative of the corporation, or is or was serving at the request or for the benefit of the Company as a director, officer, employee, agent, partner, or fiduciary of, or in any other capacity for, another corporation or any partnership, joint venture, trust, employee benefit plan or other enterprise, shall be indemnified by the Company for any losses or expenses incurred in connection with service as an officer or director of the Company, if the director or officer acted in good faith and in a manner he reasonably believed to be in, or not opposed to, the best interests of the Company and, with respect to any criminal proceeding, had no reasonable cause to believe his conduct was unlawful.
Pennsylvania law requires that to the extent that a director or officer of the Company has been successful on the merits or otherwise in defense of any action or proceeding referred to above or in defense of any claim, issue or matter therein, that director or officer shall be indemnified against expenses (including attorney fees) actually and reasonably incurred by him in connection therewith. The Company’s by-laws further provide that the right to indemnification includes the right to have expenses reasonably incurred in defending any action or proceeding described above paid by the Company in advance of the final disposition of the action or proceeding to the fullest extent permitted by Pennsylvania law; provided that, if required by Pennsylvania law, the payment of such expenses incurred in advance of the final disposition of the action or proceeding shall be made only upon delivery to the Company of an undertaking to repay all amounts so advanced without interest if it is ultimately determined that the director or officer is not entitled to be indemnified.
Indemnification shall not be made in respect of any claim, issue or matter as to which the person has been adjudged to be liable to the Company unless and only to the extent that a court determines that, despite the adjudication of liability but in view of all the circumstances of the case, the person is fairly and reasonably entitled to indemnity for the expenses that the court deems proper. Nor shall indemnification be made in any case where the act or failure to act giving rise to the claim for indemnification is determined by a court to have constituted willful misconduct or recklessness.
II-1
Item 15. Recent Sales of Unregistered Securities.
Private Placement
On July 7, 2015, the Company closed a Private Placement with certain accredited investors in which it sold 1,379,311 shares of common stock at a price per share of $11.60, for net proceeds of approximately $14.8 million. The Company paid the placement agents a fee equal to 6.0% of the aggregate gross proceeds from the Private Placement, plus reimbursement of certain expenses. The shares of common stock sold in the Private Placement were sold in reliance on the exemption from registration afforded by Section 4(a)(2) of the Securities Act and Rule 506 of Regulation D promulgated under the Securities Act. Each of the investors represented that it was acquiring the shares for investment only and not with a view towards, or for resale in connection with, the public sale or distribution thereof.
Alkermes and Orbimed Warrants
On April 10, 2015, in connection with the closing of the Company’s acquisition from Alkermes Pharma Ireland Limited (“APIL”) and pursuant to a Purchase and Sale Agreement dated March 7, 2015 (the “Purchase and Sale Agreement”), the Company issued to APIL a warrant to purchase 350,000 shares of the Company’s common stock at an exercise price equal to $19.46 per share (the “Alkermes Warrant”). On April 10, 2015, the Company also issued to OrbiMed Royalty Opportunities II, LP (“Orbimed”), pursuant to the Company’s Credit Agreement with Orbimed (the “Credit Agreement”), a warrant to purchase 294,928 shares of the Company’s common stock at a price equal to $3.28 per share, subject to certain adjustments (the “Orbimed Warrant, and together with the Alkermes Warrant, the “Warrants”).
The Warrants are exercisable until the April 10, 2022. The number of shares for which the Warrants are exercisable and the associated exercise prices are subject to certain adjustments as set forth in the Warrants. The holders of the Warrants have the right to net exercise any outstanding Warrants for shares of common stock. As specified in each of the Warrants, upon a change of control of the Company, to the extent that the Warrants are not assumed by the acquiring entity or, in the case of the Orbimed Warrant, automatically exercised, the holder can elect to receive, subject to certain limitations and assumptions, cash equal to the Black-Scholes value of the outstanding Warrants.
The Company relied on the exemption from registration contained in Section 4(2) of the Securities Act, and Regulation D, Rule 506 thereunder, for the issuance of the Warrants and the shares of Common Stock issuable pursuant to such Warrants (the “Warrant Shares”). As part of executing the Purchase and Sale Agreement and the Credit Agreement and receiving the Warrants and the Warrant Shares, Orbimed and each of the Sellers each represented that it is an “accredited investor” as defined in Regulation D of the Securities Act and that the securities purchased by them will be acquired solely for their own account for investment and not with a view to or for sale or distribution of the Warrants or the Warrant Shares or any part thereof.
Aspire Capital Transaction
On February 2, 2015, the Company entered into a Common Stock Purchase Agreement, or the Purchase Agreement, with Aspire Capital Fund, LLC, an Illinois limited liability company, or Aspire Capital, which provides that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase up to an aggregate of $10.0 million of shares of the Company’s common stock (the “Purchase Shares”) over the 24-month term of the Purchase Agreement.
Upon execution of the Purchase Agreement, the Company issued 96,463 shares of its common stock to Aspire Capital in consideration for entering into the Purchase Agreement. The Purchase Shares may be sold by the Company to Aspire Capital on any business day the Company selects in two ways: (1) through a regular purchase of up to 50,000 shares at a known price based on the market price of the Company’s common stock prior to the time of each sale, and (2) through a VWAP purchase of a number of shares up to 30% of the volume traded on the purchase date at a price equal to the lessor of the
II-2
closing sale price or 95% of the volume weighted average price for such purchase date. As of November 30, 2016, the Company had issued 1,143,940 Purchase Shares to Aspire Capital under the Purchase Agreement for net proceeds of approximately $7.8 million.
The issuance of the Commitment Shares and all other shares of common stock that have been issued, or may be issued from time to time, to Aspire Capital under the Purchase Agreement is exempt from registration under the Securities Act of 1933, as amended (the “Securities Act”), pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(a)(2) of the Securities Act.
IPO Warrants
Upon the closing of the IPO, the Company issued to Aegis Capital Corporation, the representative of the underwriters for the IPO, warrants to purchase up to 150,000 shares of the Company’s common stock, with a per share exercise price equal to $12.00, or 150% of the public offering price. The warrants are exercisable by the underwriters at any time, in whole or in part, until March 12, 2019. On July 22, 2015, the Company issued 2,941 shares of its common stock pursuant to a cashless exercise of 10,000 IPO warrants with a per share exercise price of $12.00 per share .The issuance of the IPO warrants and all other shares of common stock that have been and may be issued upon exercise of the IPO warrants is exempt from registration under the Securities Act pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(a)(2) of the Securities Act.
Conversion of Series A Redeemable Convertible Preferred Stock
Upon the closing of the IPO on March 12, 2014, all shares of the Company’s then-outstanding Series A Redeemable Convertible Preferred Stock and all of the Company’s then-outstanding 8% Convertible Promissory Notes automatically converted into an aggregate of 3,239,500 shares of common stock. The issuance qualified for exemption under Section 3(a) (9) of the Securities Act.
8% Convertible Promissory Notes
In each of calendar years 2012, 2013, and 2014, the Company sold an aggregate of $0.9 million, $0.7 million, and $0.1 million, respectively, of its 8% Convertible Promissory Notes in private placements to SCP Vitalife Partners II, L.P., and $0.3 million, $0.2 million, and $0.05 million, respectively, of its 8% Convertible Promissory Notes in private placements to SCP Vitalife Partners (Israel) II, L.P. The notes accrued interest at a rate of 8% compounded quarterly. The notes converted into shares of the Company’s common stock at the IPO. The notes were issued to SCP Vitalife Partners II, L.P. and SCP Vitalife Partners (Israel) II, L.P. in reliance upon the exemption from the registration requirements of the Securities Act, as set forth in Section 4(a)(2) of the Securities Act relative to transactions by an issuer not involving a public offering. All purchasers of the 8% Convertible Promissory Notes represented to the Company in connection with their purchase that they were accredited investors and were acquiring such notes for their own account for investment purposes only and not with a view to, or for sale in connection with, any distribution thereof and that they could bear the risks of the investment and could hold the securities for an indefinite period of time. The purchasers received written disclosures that the securities had not been registered under the Securities Act and that any resale must be made pursuant to a registration statement or an available exemption from such registration.
Stock Option Grants
Through March 2014, prior to the Company’s IPO and prior to the effectiveness of the Company’s registration statement on Form S-8, the Company granted stock options under the 2008 Stock Option Plan and the 2013 Equity Incentive Plan to purchase an aggregate of 334,800 shares of the Company’s common stock at a weighted-average exercise price of $6 per share to certain directors, employees and consultants. The stock options and the common stock issuable upon the exercise of such options were
II-3
issued pursuant to written compensatory plans or arrangements with the Company’s directors, employee and consultants, in reliance on the exemption from the registration requirements of the Securities Act provided by Rule 701 promulgated under the Securities Act or the exemption set forth in Section 4(a)(2) under the Securities Act relative to transactions by an issuer not involving any public offering. All recipients either received adequate information about us or had access through the Company’s employment or other relationships to such information.
Item 16. Exhibits.
See the Exhibit Index on the page immediately preceding the exhibits for a list of exhibits filed as part of this Registration Statement on Form S-1, which Exhibit Index is incorporated herein by reference.
Item 17. Undertakings.
(b) The undersigned registrant hereby undertakes that, for purposes of determining any liability under the Securities Act of 1933, each filing of the registrant’s annual report pursuant to section 13(a) or section 15(d) of the Securities Exchange Act of 1934 (and, where applicable, each filing of an employee benefit plan’s annual report pursuant to section 15(d) of the Securities Exchange Act of 1934) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(e) The undersigned registrant hereby undertakes to deliver or cause to be delivered with the prospectus, to each person to whom the prospectus is sent or given, the latest annual report to security holders that is incorporated by reference in the prospectus and furnished pursuant to and meeting the requirements of Rule 14a-3 or Rule 14c-3 under the Securities Exchange Act of 1934; and, where interim financial information required to be presented by Article 3 of Regulation S-X are not set forth in the prospectus, to deliver, or cause to be delivered to each person to whom the prospectus is sent or given, the latest quarterly report that is specifically incorporated by reference in the prospectus to provide such interim financial information.
(h) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
(i) The undersigned registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b) (1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-4
SIGNATURES
Pursuant to the requirements of the Securities Act, the Registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-1 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in Malvern, Pennsylvania, on December 1, 2016.
| RECRO PHARMA, INC. | ||
| By: |
/s/ GERRI A. HENWOOD | |
| Gerri A. Henwood Chief Executive Officer | ||
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below appoints Gerri A. Henwood and Michael Celano, and each of them acting individually, and each of whom may act without the joinder of the other, as his or her true and lawful attorneys-in-fact and agents, with full power of each to act alone, with full powers of substitution and resubstitution for him or her and in his or her name, place and stead, in any and all capacities, to sign this Registration Statement on Form S-1 for Recro Pharma, Inc., along with any or all amendments (including post-effective amendments) to this Registration Statement, and any other registration statements for the same offering pursuant to Rule 462(b) of the Securities Act of 1933, as amended, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, with full power of each to act alone, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or would do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or his substitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, as amended, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ GERRI A. HENWOOD Gerri A. Henwood |
President, Chief Executive Officer and Director (Principal Executive Officer) |
December 1, 2016 | ||
| /s/ MICHAEL CELANO Michael Celano |
Chief Financial Officer (Principal Financial Officer) |
December 1, 2016 | ||
| /s/ DONNA NICHOLS Donna Nichols |
Vice President, Corporate Controller (Principal Accounting Officer) |
December 1, 2016 | ||
| /s/ ALFRED ALTOMARI Alfred Altomari |
Director |
December 1, 2016 | ||
| /s/ WILLIAM L. ASHTON William L. Ashton |
Director |
December 1, 2016 | ||
| /s/ MICHAEL BERELOWITZ Michael Berelowitz |
Director |
December 1, 2016 | ||
II-5
| Signature |
Title |
Date | ||
| /s/ WINSTON J. CHURCHILL Winston J. Churchill |
Director |
December 1, 2016 | ||
| /s/ KAREN FLYNN Karen Flynn |
Director |
December 1, 2016 | ||
| /s/ ABRAHAM LUDOMIRSKI Abraham Ludomirski |
Director |
December 1, 2016 | ||
| /s/ WAYNE B. WEISMAN Wayne B. Weisman |
Director |
December 1, 2016 | ||
II-6
EXHIBIT INDEX
| Exhibit |
Description |
Method of Filing | ||
| 1.1 | Form of Underwriting Agreement. | Filed herewith. | ||
| 2.1† | Purchase and Sale Agreement, dated March 7, 2015, by and among Recro Pharma, Inc., Recro Pharma LLC, Daravita Limited, Alkermes Pharma Ireland Limited and Eagle Holdings USA, Inc. | Incorporated herein by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on March 11, 2015. | ||
| 3.1 | Second Amended and Restated Articles of Incorporation of Recro Pharma, Inc. | Incorporated herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on March 13, 2014. | ||
| 3.2 | Third Amended and Restated Bylaws of Recro Pharma, Inc. | Incorporated herein by reference to Exhibit 3.2 to the Company’s Current Report on Form 8-K filed on March 13, 2014. | ||
| 4.1 | Specimen certificate evidencing shares of common stock. | Incorporated herein by reference to Exhibit 4.1 to the Company’s Registration Statement on Form S-1/A filed on December 20, 2013. | ||
| 4.2 | Investor Rights Agreement, dated September 4, 2008, by and among Recro Pharma, Inc., and the investors party thereto. | Incorporated herein by reference to Exhibit 4.2 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 4.3 | Registration Rights Agreement, dated February 2, 2015, between Recro Pharma, Inc. and Aspire Capital Fund, LLC. | Incorporated herein by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on February 3, 2015. | ||
| 4.4 | Form of Alkermes Warrant. | Incorporated herein by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on March 11, 2015. | ||
| 4.5 | Form of OrbiMed Warrant. | Incorporated herein by reference to Exhibit 4.2 to the Company’s Current Report on Form 8-K filed on March 11, 2015. | ||
| 4.6 | Form of IPO Warrant. | Incorporated herein by reference to Exhibit A of Exhibit 1.1 to the Company’s Registration Statement on Form S-1/A filed on February 11, 2014. | ||
| 5.1 | Form of Opinion of Hogan Lovells US LLP. | Filed herewith. | ||
| 10.1† | Dexmedetomidine License Agreement, dated August 22, 2008, by and among Recro Pharma, Inc. and Orion Corporation. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.2† | First Amendment to Dexmedetomidine License Agreement, dated January 17, 2009, by and between Recro Pharma, Inc., and Orion Corporation. | Incorporated herein by reference to Exhibit 10.2 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.3† | Dexmedetomidine API Supply Agreement, dated August 22, 2008, by and among Recro Pharma, Inc., and Orion Corporation. | Incorporated herein by reference to Exhibit 10.3 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
II-7
| Exhibit |
Description |
Method of Filing | ||
| 10.4† | Fadolmidine License Agreement, dated July 21, 2010, by and among Recro Pharma, Inc. and Orion Corporation. | Incorporated herein by reference to Exhibit 10.4 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.5• | Employment Agreement, dated October 8, 2013, between Recro Pharma, Inc. and Gerri Henwood. | Incorporated herein by reference to Exhibit 10.5 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.6• | Separation and Mutual Release Agreement, dated December 8, 2015, between Recro Pharma, Inc. and Charles Garner. | Incorporated herein by reference to Exhibit 10.6 to the Company’s Post-Effective Amendment on Form S-1 filed on December 23, 2015. | ||
| 10.7• | Employment Agreement, dated October 9, 2013, between Recro Pharma, Inc. and Randall Mack. | Incorporated herein by reference to Exhibit 10.7 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.8• | Employment Agreement, dated October 9, 2013, between Recro Pharma, Inc. and Diane Myers. | Incorporated herein by reference to Exhibit 10.8 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.9• | Employment Agreement, dated October 9, 2013, between Recro Pharma, Inc. and Donna Nichols. | Incorporated herein by reference to Exhibit 10.9 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.10• | Employment Agreement, dated December 1, 2015, between Recro Pharma, Inc. and Stewart McCallum, M.D. | Incorporated herein by reference to Exhibit 10.33 to the Company’s Post-Effective Amendment on Form S-1 filed on December 23, 2015. | ||
| 10.11• | Employment Agreement, dated December 1, 2015, between Recro Pharma, Inc. and Fred Graff. | Incorporated herein by reference to Exhibit 10.11 to the Company’s Annual Report on Form 10-K filed on March 24, 2016. | ||
| 10.12• | Employment Agreement, dated July 1, 2016, between Recro Pharma, Inc. and Michael Celano. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on November 10, 2016. | ||
| 10.13• | 2008 Stock Option Plan. | Incorporated herein by reference to Exhibit 10.10 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.14• | Form of 2008 Stock Option Plan Award Agreement. | Incorporated herein by reference to Exhibit 10.11 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.15• | 2013 Equity Incentive Plan. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on March 13, 2014. | ||
| 10.16• | Form of 2013 Equity Incentive Plan Award Agreement. | Incorporated herein by reference to Exhibit 10.14 to the Company’s Annual Report on Form 10-K filed on March 25, 2015. | ||
II-8
| Exhibit |
Description |
Method of Filing | ||
| 10.17• | Recro Pharma, Inc. Amended and Restated Equity Incentive Plan | Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on June 26, 2015. | ||
| 10.18• | Form of Recro Pharma, Inc. Equity Incentive Plan Award Agreement. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on December 22, 2015. | ||
| 10.19• | Form of Recro Pharma, Inc. Equity Incentive Plan Award Agreement Award Agreement for Restricted Stock Units. | Incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on December 22, 2015. | ||
| 10.20• | Form of Award Agreement for Inducement Awards. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Registration Statement on Form S-8 filed on December 23, 2015. | ||
| 10.21 | Master Consulting Services Agreement, dated October 10, 2013, by and between Recro Pharma, Inc. and Malvern Consulting Group, Inc. | Incorporated herein by reference to Exhibit 10.14 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.22 | Office Services Agreement, dated January 1, 2012, between Recro Pharma, Inc. and Malvern Consulting Group, Inc. | Incorporated herein by reference to Exhibit 10.15 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.23 | Amendment #1 to the Office Services Agreement, dated October 3, 2013, by and between Recro Pharma, Inc. and Malvern Consulting Group, Inc. | Incorporated herein by reference to Exhibit 10.16 to the Company’s Registration Statement on Form S-1/A filed on November 29, 2013. | ||
| 10.24 | Common Stock Purchase Agreement, dated February 2, 2015, between Recro Pharma, Inc. and Aspire Capital Fund, LLC. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on February 3, 2015. | ||
| 10.25† | Credit Agreement, dated as of March 7, 2015, by and between Recro Pharma LLC and OrbiMed Royalty Opportunities II, LP. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on March 11, 2015. | ||
| 10.26 | First Amendment to Credit Agreement, dated as of April 10, 2015, by and between Recro Pharma LLC and OrbiMed Royalty Opportunities II, LP. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on April 16, 2015. | ||
| 10.27 | Fifth Amendment to Credit Agreement, dated as of November 12, 2015, by and between Recro Pharma LLC and OrbiMed Royalty Opportunities II, LP. | Incorporated herein by reference to Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q filed on November 13, 2015. | ||
| 10.28 | Guarantee, dated as of March 7, 2015, by Recro Pharma, Inc. in favor of OrbiMed Royalty Opportunities II, LP. | Incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on March 11, 2015. | ||
II-9
| Exhibit |
Description |
Method of Filing | ||
| 10.29† | Asset Transfer and License Agreement, dated as of April 10, 2015, between Alkermes Pharma Ireland Limited and DV Technology LLC. | Incorporated herein by reference to Exhibit 10.5 to the Company’s Quarterly Report on Form 10-Q filed on May 12, 2015. | ||
| 10.30† | Transition Services Agreement, dated as of April 10, 2015, by and among Alkermes Pharma Ireland Limited, Recro Pharma, Inc., DV Technology LLC, and Alkermes Gainesville LLC. | Incorporated herein by reference to Exhibit 10.6 to the Company’s Quarterly Report on Form 10-Q filed on May 12, 2015. | ||
| 10.31† | Development, Manufacturing and Supply Agreement, dated July 10, 2015, by and between Alkermes Pharma Ireland Limited and Recro Pharma, Inc. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on August 14, 2015 | ||
| 10.32† | Amended and Restated License and Supply Agreement, dated June 26, 2003, by and among Elan Corporation, plc (predecessor-in-interest to Recro Gainesville LLC) and Watson Laboratories, Inc. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on August 14, 2015 | ||
| 10.33 | Supplemental Agreement, dated December 8, 2004, to Amended and Restated License and Supply Agreement, dated June 26, 2003, by and among Elan Corporation, plc (predecessor-in-interest to Recro Gainesville LLC) and Watson Laboratories, Inc. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on August 14, 2015 | ||
| 10.34 | Supplemental Agreement No. 2, dated January 17, 2014, to Amended and Restated License and Supply Agreement, dated June 26, 2003, by and among Elan Corporation, plc (predecessor-in-interest to Recro Gainesville LLC) and Watson Laboratories, Inc. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on August 14, 2015 | ||
| 10.35 | Form of Securities Purchase Agreement, dated July 1, 2015, by and among Recro Pharma, Inc. and the Purchasers party thereto. | Incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on July 8, 2015 | ||
| 21.1 | Subsidiaries of Recro Pharma, Inc. | Incorporated herein by reference to Exhibit 21.1 to the Company’s Annual Report on Form 10-K filed on March 24, 2016. | ||
| 23.1 | Consent of KPMG LLP, Independent Registered Public Accounting Firm. | Filed herewith. | ||
| 23.2 | Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm | Filed herewith. | ||
II-10
| Exhibit |
Description |
Method of Filing | ||
| 23.3 | Consent of Hogan Lovells US LLP (included in Exhibit 5.1) | |||
| 24.1 | Power of Attorney. | Included on the signature page hereto. | ||
| * | To be filed by amendment. |
| • | Exhibit relates to compensation arrangements. |
| † | Confidential treatment has been granted with respect to certain portions of this exhibit. Omitted portions have been filed with the Securities and Exchange Commission. |
II-11